Aktīvās sastāvdaļas: sunitinibs
SUTENT 12,5 mg cietās kapsulas
SUTENT 25 mg cietās kapsulas
SUTENT 37,5 mg cietās kapsulas
SUTENT 50 mg cietās kapsulas
Indikācijas Kāpēc lieto Sutent? Kam tas paredzēts?
Sutent satur aktīvo vielu sunitinibu, kas ir proteīnkināzes inhibitors. To lieto vēža ārstēšanai, novēršot noteiktas olbaltumvielu grupas, kas, kā zināms, ir iesaistīta vēža šūnu augšanā un izplatībā, aktivitāti.
Sutent Jums parakstīs tikai ārsts, kuram ir pieredze pretvēža zāļu lietošanā.
Sutent lieto, lai ārstētu pieaugušos ar šādiem vēža veidiem:
- Kuņģa -zarnu trakta stromas vēzis (GIST), kuņģa un zarnu vēža veids, gadījumos, kad imatinibs (citas pretvēža zāles) vairs nedarbojas vai to vairs nevar lietot.
- Metastātisks nieru vēzis (MRCC), nieru vēža veids, kas izplatījies uz citām ķermeņa daļām.
- Aizkuņģa dziedzera neiroendokrīnie audzēji (pNET) (aizkuņģa dziedzera hormonu ražojošo šūnu audzēji), kas progresē vai nav noņemami
. Ja neesat pārliecināts, kā Sutent darbojas vai kāpēc šīs zāles ir parakstītas Jums, jautājiet savam ārstam.
Kontrindikācijas Kad Sutent nedrīkst lietot
Nelietojiet Sutent šādos gadījumos:
- Ja Jums ir alerģija pret sunitinibu vai kādu citu (6. punktā minēto) šo zāļu sastāvdaļu.
Piesardzība lietošanā Kas jāzina pirms Sutent lietošanas
Pirms Sutent lietošanas pastāstiet ārstam:
- Ja Jums ir augsts asinsspiediens. Sutent var paaugstināt asinsspiedienu. Ārsts var pārbaudīt asinsspiedienu Sutent lietošanas laikā un nepieciešamības gadījumā viņam būs jālieto zāles asinsspiediena pazemināšanai.
- Ja Jums ir vai ir bijušas asins slimības, asiņošanas problēmas vai sasitumi. Ārstēšana ar Sutent var izraisīt paaugstinātu asiņošanas risku, dažu asins šūnu skaita izmaiņas, kuru trūkums izraisa anēmiju vai ietekmē asins recēšanas spēju. Asiņošanas risks var būt lielāks, ja lietojat varfarīnu vai acenokumarolu - zāles, kas šķidrina asinis, lai novērstu asins recekļu veidošanos. Pastāstiet ārstam, ja Sutent lietošanas laikā rodas asiņošana.
- Ja Jums ir sirds problēmas. Sutent var izraisīt sirds problēmas. Pastāstiet ārstam, ja jūtaties ļoti noguris, elpas trūkums vai pietūkušas pēdas un potītes.
- Ja Jums rodas patoloģiskas sirds ritma izmaiņas. Sutent var izraisīt izmaiņas sirds ritmā. Kamēr ārstējaties ar Sutent, ārsts var Jums veikt elektrokardiogrammu, lai novērtētu šo izmaiņu apmēru. Pastāstiet savam ārstam, ja ārstēšanas laikā ar Sutent jūtat reiboni, ģīboni vai sirdsdarbības traucējumus.
- Ja Jums nesen ir bijušas problēmas ar asins recekļu veidošanos vēnās un / vai artērijās (asinsvadu tipi), ieskaitot insultu, sirdslēkmi, emboliju vai trombozi. Nekavējoties sazinieties ar savu ārstu, ja Jums rodas simptomi, piemēram, sasprindzinājums krūtīs vai sāpes, sāpes rokās, mugurā, kaklā vai žoklī, elpas trūkums, nejutīgums vai vājums vienā ķermeņa pusē, trīcoša staigāšana, sāpes ārstēšanas ar Sutent laikā. vai reibonis.
- Ja Jums ir problēmas ar vairogdziedzeri. Sutent var izraisīt vairogdziedzera darbības traucējumus. Pastāstiet savam ārstam, ja Sutent lietošanas laikā esat noguris vieglāk, parasti jūtaties vēsāks nekā citi cilvēki vai arī jūsu balss samazinās. Pirms Sutent lietošanas un zāļu lietošanas laikā regulāri jāpārbauda vairogdziedzera darbība. Ja vairogdziedzeris neražo pietiekami daudz vairogdziedzera hormonu, var būt nepieciešams lietot vairogdziedzera hormona aizstājēju.
- Ja Jums ir vai ir bijušas aizkuņģa dziedzera vai žultspūšļa problēmas. Pastāstiet ārstam, ja Jums rodas kāda no šīm pazīmēm un simptomiem: sāpes kuņģī (vēdera augšdaļā), slikta dūša, vemšana un drudzis.Tos var izraisīt "aizkuņģa dziedzera vai žultspūšļa iekaisums.
- Ja Jums ir vai kādreiz ir bijuši aknu darbības traucējumi. Pastāstiet ārstam, ja ārstēšanas laikā ar Sutent Jums rodas kāda no šīm aknu darbības pazīmēm un simptomiem: nieze, ādas vai acu dzelte, tumšs urīns un sāpes vai diskomforts vēdera augšējā labajā daļā. veikt testus, lai pārbaudītu aknu darbību pirms ārstēšanas ar Sutent un tās laikā, kā arī klīniski piemērotos gadījumos.
- Ja Jums ir vai ir bijuši nieru darbības traucējumi. Ārsts uzraudzīs nieru darbību.
- Ja Jums ir paredzēta operācija vai nesen veikta operācija. Sutent var ietekmēt jūsu brūču dzīšanu. Parasti, ja jūs gatavojaties operācijai, Jums jāpārtrauc Sutent lietošana. Ārsts izlems, kad atsākt ārstēšanu ar Sutent.
- Pirms ārstēšanas uzsākšanas ar Sutent ieteicams pārbaudīt zobus.
- ja Jums ir vai ir bijušas sāpes mutē, zobos un / vai žoklī, pietūkums vai čūlas mutē, nejutīgums vai smaguma sajūta žoklī vai zobu atslābināšanās, nekavējoties pastāstiet par to savam ārstam un zobārstam.
- ja Jums tiek veikta invazīva zobu ārstēšana vai zobu ķirurģija, lūdzu, pastāstiet savam ārstam, ka tiekat ārstēts ar Sutent, īpaši, ja lietojat arī intravenozus bisfosfonātus vai esat tos lietojis iepriekš. Bifosfonāti ir zāles, ko lieto, lai novērstu kaulu komplikācijas, kuras varētu būt parakstītas citas medicīniskas problēmas dēļ.
- Ja Jums ir vai kādreiz ir bijuši ādas un zemādas audu bojājumi. Ārstēšanas laikā ar šīm zālēm var rasties „gangrenozā pioderma” (sāpīga ādas čūla) vai „nekrotizējošs fascīts” (ātri izplatīga „ādas / mīksto audu infekcija, kas var būt letāla). Ārstēšanas pārtraukšana. Nopietnas ādas reakcijas (Stīvenss-Džonsons) sindroms, toksiska epidermas nekrolīze, multiformā eritēma), lietojot sunitinibu, sākotnēji parādās uz stumbra kā sarkanīgi mērķa formas plankumi vai apļveida plankumi, bieži ar pūslīšiem centrā. Reakcija var progresēt līdz plaši izplatītiem pūslīšiem vai ādas lobīšanai, un tā var būt letāla. Ja Jums rodas izsitumi vai kāds no šiem ādas simptomiem, nekavējoties apmeklējiet ārstu.
- Ja Jums ir vai ir bijuši krampji. Pēc iespējas ātrāk pastāstiet ārstam, ja Jums ir paaugstināts asinsspiediens, galvassāpes, redzes zudums.
- Ja Jums ir diabēts. Cukura diabēta slimniekiem regulāri jāpārbauda cukura līmenis asinīs, lai noskaidrotu, vai ir jāmaina cukura diabēta zāļu deva, lai samazinātu zemu cukura līmeni asinīs.
Bērni un pusaudži
Sutent nav indicēts pacientiem līdz 18 gadu vecumam. Sutent nav pētīts bērniem un pusaudžiem.
Mijiedarbība Kādas zāles vai pārtikas produkti var mainīt Sutent iedarbību
Pastāstiet ārstam vai farmaceitam par visām zālēm, kuras lietojat, pēdējā laikā esat lietojis vai varētu lietot, ieskaitot zāles, kas iegādātas bez receptes un bez receptes.
Dažas zāles var mainīt Sutent līmeni organismā. Jums jāinformē ārsts, ja lietojat zāles, kas satur šādas aktīvās vielas:
- ketokonazolu, itrakonazolu - lieto sēnīšu infekciju ārstēšanai
- eritromicīnu, klaritromicīnu, rifampicīnu - lieto infekciju ārstēšanai
- ritonavīrs - lieto AIDS ārstēšanai
- deksametazons - kortikosteroīds, ko lieto vairākiem apstākļiem
- fenitoīns, karbamazepīns, fenobarbitāls - lieto epilepsijas un citu neiroloģisku slimību ārstēšanai
- augu izcelsmes preparāti, kas satur asinszāli (Hypericum perforatum) - lieto depresijas un trauksmes ārstēšanai
Sutent kopā ar uzturu un dzērienu
Ārstēšanas laikā ar Sutent jāizvairās no greipfrūtu sulas uzņemšanas.
Brīdinājumi Ir svarīgi zināt, ka:
Grūtniecība un zīdīšanas periods
Ja esat grūtniece vai jums ir aizdomas par grūtniecību, lūdzu, pastāstiet to ārstam.
Sutent nedrīkst lietot grūtniecības laikā, ja vien tas nav absolūti nepieciešams. Jūsu ārsts apspriedīs ar Jums iespējamos Sutent terapijas riskus grūtniecības laikā.
Ja grūtniecība ir iespējama, ārstēšanas laikā ar Sutent jāizmanto droša kontracepcijas metode.
Ja barojat bērnu ar krūti, lūdzu, pastāstiet to ārstam. Ārstēšanas laikā ar Sutent nedrīkst barot bērnu ar krūti.
Transportlīdzekļu vadīšana un mehānismu apkalpošana
Ja jūtat reiboni vai neparasti nogurušu, esiet īpaši piesardzīgs, vadot transportlīdzekli vai apkalpojot mehānismus.
Deva, lietošanas veids un laiks Kā lietot Sutent: Devas
Vienmēr lietojiet šīs zāles tieši tā, kā ārsts Jums stāstījis.
Ja rodas šaubas, konsultējieties ar ārstu. Ārsts izrakstīs Jums pareizo devu, pamatojoties uz vēža veidu, kas Jums jāārstē. Ja Jums ārstē GIST vai MRCC, parastā deva ir 50 mg vienu reizi dienā, kas jālieto 28 dienas (4 nedēļas), kam seko 14 dienu (2 nedēļu) atpūta (bez medikamentiem) 6 nedēļu ciklos. Ja Jums tiek ārstēts pNET, parastā deva ir 37,5 mg vienu reizi dienā bez pārtraukuma. Ārsts noteiks nepieciešamo devu un to, kad pārtraukt ārstēšanu ar Sutent. Sutent var lietot kopā ar ēdienu vai bez tā.
Pārdozēšana Ko darīt, ja esat lietojis pārāk daudz Sutent
Ja esat lietojis Sutent vairāk nekā noteikts
Ja nejauši esat lietojis pārāk daudz kapsulu, nekavējoties konsultējieties ar ārstu. Var būt nepieciešama medicīniska palīdzība
Ja esat aizmirsis lietot Sutent
Nelietojiet dubultu devu, lai aizvietotu aizmirsto devu.
Blakusparādības Kādas ir Sutent blakusparādības
Tāpat kā citas zāles, šīs zāles var izraisīt blakusparādības, kaut arī ne visiem tās izpaužas.
Nekavējoties sazinieties ar savu ārstu, ja Jums rodas kāda no šīm nopietnajām blakusparādībām (skatīt arī Kas jāzina pirms Sutent lietošanas):
Sirds problēmas. Pastāstiet ārstam, ja jūtaties ļoti noguris, elpas trūkums vai pietūkušas pēdas un potītes. Tie varētu būt sirdsdarbības traucējumu simptomi, piemēram, sirds mazspēja un sirds muskuļu problēmas (kardiomiopātija).
Plaušu vai elpošanas problēmas. Pastāstiet ārstam, ja Jums rodas klepus, sāpes krūtīs, pēkšņs elpas trūkums vai asiņu klepus. Tie varētu būt plaušu embolijas simptomi, kas rodas, kad asins recekļi nonāk plaušās.
Nieru problēmas. Pastāstiet ārstam, ja Jums ir “mainīts urinēšanas biežums vai prombūtne, kas varētu būt nieru mazspējas simptomi”.
Asiņošana. Nekavējoties pastāstiet ārstam, ja Sutent lietošanas laikā rodas kāds no šiem simptomiem vai nopietna asiņošanas problēma: pietūkušas, sāpīgas vēdera daļas (vēders); vemšana ar asinīm; tumši, lipīgi izkārnījumi; galvassāpes vai garīgā stāvokļa izmaiņas, asiņu vai krēpu klepus ar asinīm no plaušām vai elpceļiem.
Audzēja iznīcināšana, kas izraisa zarnu perforāciju.Pastāstiet savam ārstam, ja Jums ir stipras sāpes zarnās, drudzis, slikta dūša, vemšana, asinis izkārnījumos vai izmaiņas zarnu paradumos.
Citas blakusparādības, kas var rasties, lietojot Sutent, ir:
Ļoti bieži sastopamas blakusparādības (var skart vairāk nekā 1 no 10 cilvēkiem)
- Trombocītu, sarkano asins šūnu un / vai balto asins šūnu (piemēram, neitrofilu) skaita samazināšanās.
- Elpas trūkums
- Augsts asinsspiediens.
- Pārmērīgs nogurums, spēka zudums.
- Pietūkums, ko izraisa šķidrums zem ādas un ap acīm, dziļi alerģiski izsitumi.
- Sāpes / kairinājums mutē, sāpīgums / iekaisums / sausa mute, garšas traucējumi, kuņģa darbības traucējumi, slikta dūša, vemšana, caureja, aizcietējums, sāpes vēderā / pietūkums, apetītes zudums / samazināšanās.
- Samazināta vairogdziedzera darbība (hipotireoze).
- Reibonis
- Galvassāpes.
- Deguna asiņošana.
- Muguras sāpes, locītavu sāpes.
- Sāpes rokās un kājās.
- Ādas dzeltēšana / ādas krāsas maiņa, pārmērīga ādas pigmentācija, matu krāsas izmaiņas, izsitumi uz plaukstām un pēdām, izsitumi, sausa āda.
- Klepus.
- Drudzis.
- Grūtības aizmigt.
Biežas blakusparādības (var skart 1 līdz 10 no 100 cilvēkiem)
- Trombu veidošanās asinsvados.
- Nepietiekama asins piegāde sirds muskuļiem koronāro artēriju obstrukcijas vai sašaurināšanās dēļ.
- Sāpes krūtīs.
- Samazināts asins daudzums, ko sūknē sirds.
- Šķidruma aizture arī ap plaušām.
- Infekcijas.
- Samazināts cukura līmenis asinīs. Ja Jums rodas zema cukura līmeņa asinīs pazīmes un simptomi: pēc iespējas ātrāk informējiet ārstu, ja Jums rodas nogurums, sirdsklauves, svīšana, izsalkums un samaņas zudums.
- Olbaltumvielu zudums urīnā, kas dažkārt izraisa pietūkumu.
- Gripas sindroms.
- Nenormāli asins analīzes, ieskaitot aknu un aizkuņģa dziedzera enzīmu līmeni.
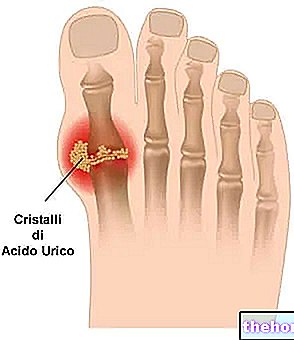
- Augsts urīnskābes līmenis asinīs.
- Hemoroīdi, sāpes taisnās zarnās, smaganu asiņošana, apgrūtināta rīšana vai nespēja norīt.
- Dedzinoša vai sāpīga sajūta mēlē, gremošanas trakta gļotādas iekaisums, gāzu pārpalikums kuņģī vai zarnās.
- Svara zudums.
- Skeleta -muskuļu sāpes (sāpes muskuļos un kaulos), muskuļu vājums, muskuļu nogurums, muskuļu sāpes, muskuļu spazmas.
- Deguna sausums, aizlikts deguns.
- Pārmērīga asarošana.
- Ādas jutīguma izmaiņas, sausa āda, nieze, lobīšanās un ādas iekaisums, tulznas, pūtītes, nagu krāsas izmaiņas, matu izkrišana.
- Nenormālas sajūtas ekstremitātēs.
- Pārmērīga jutības samazināšanās / palielināšanās, īpaši pieskaroties.
- Dedzināšana kuņģī.
- Dehidratācija.
- Sejas apsārtums.
- Izmaiņas urīna krāsā.
- Depresija.
- Drebuļi.
Retākas blakusparādības (var skart 1 līdz 10 no 1000 cilvēkiem)
- Mīksto audu infekcijas, arī anoģenitālā reģionā, potenciāli dzīvībai bīstamas. Nekavējoties sazinieties ar savu ārstu, ja Jums rodas infekcijas simptomi ap ādas brūci, tostarp drudzis, sāpes, apsārtums, pietūkums vai strutas vai asiņu aizplūšana.
- Insults.
- Sirdslēkme, ko izraisa pārtraukta vai samazināta asins piegāde sirdij.
- Sirds elektriskās aktivitātes izmaiņas vai sirds ritma izmaiņas.
- Šķidrums ap sirdi (izsvīdums perikardā).
- Aknu mazspēja.
- Sāpes vēderā, ko izraisa "aizkuņģa dziedzera iekaisums.
- Audzēja iznīcināšana, kas izraisa zarnu perforāciju.
- Žultspūšļa iekaisums (pietūkums un apsārtums) ar vai bez saistītiem akmeņiem.
- Nenormāls saziņas kanāls starp diviem ķermeņa dobumiem vai ar ādu.
- Sāpes mutē, zobos un / vai žoklī, pietūkums vai kairinājums mutē, nejutīgums vai smaguma sajūta žoklī, vai zobu atslābums. Tās varētu būt žokļa / žokļa kaula bojājuma (osteonekrozes) pazīmes un simptomi. Nekavējoties pastāstiet ārstam un zobārstam, ja Jums rodas kāda no šīm pazīmēm un simptomiem.
- Pārmērīga vairogdziedzera hormonu ražošana, kā rezultātā palielinās vielmaiņa. Problēmas ar brūču sadzīšanu pēc operācijas.
- Muskuļu enzīmu (kreatīna fosfokināzes) palielināšanās asinīs.
- Nepiemērota un pārmērīga reakcija uz alergēniem.
Retas blakusparādības (var skart 1 līdz 10 no 10 000 cilvēkiem)
- Smagas ādas un / vai gļotādu reakcijas (Stīvensa-Džonsona sindroms, toksiska epidermas nekrolīze, multiformā eritēma).
- Audzēja līzes sindroms (TLS) - TLS ietver vielmaiņas komplikāciju kopumu, kas var rasties vēža ārstēšanas laikā. Tos izraisa skarto vēža šūnu sabrukšanas produkti, un tie var būt: slikta dūša, elpas trūkums, neregulāra sirdsdarbība, muskuļu krampji, krampji, duļķains urīns un nogurums, kas saistīts ar patoloģiskiem laboratorijas testu rezultātiem (augsts kālija, urīnskābes un fosforskābes līmenis) un zems kalcija līmenis asinīs), kas var izraisīt nieru darbības izmaiņas un akūtu nieru mazspēju.
- Nenormāls muskuļu sabrukums, kas var izraisīt nieru darbības traucējumus (rabdomiolīze).
- Smadzeņu darbības traucējumi, kas var izraisīt dažādus simptomus, piemēram, galvassāpes, apjukumu, krampjus un redzes zudumu (atgriezenisks atgriezeniskas leikoencefalopātijas sindroms).
- Sāpīgas ādas čūlas (gangrenozā piodermija).
- Aknu iekaisums (hepatīts).
- Vairogdziedzera iekaisums.
Ziņošana par blakusparādībām
Ja Jums rodas jebkādas blakusparādības, konsultējieties ar ārstu, ieskaitot visas iespējamās blakusparādības, kas nav minētas šajā instrukcijā. Jūs varat ziņot par blakusparādībām arī tieši, izmantojot V pielikumā minēto valsts ziņošanas sistēmu. Ziņojot par blakusparādībām, jūs varat palīdzēt iegūt vairāk informācijas par šo zāļu drošumu.
Derīguma termiņš un saglabāšana
- Uzglabāt šīs zāles bērniem neredzamā un nepieejamā vietā.
- Nelietot šīs zāles pēc derīguma termiņa beigām, kas norādīts uz kastītes un marķējuma pēc „Derīgs līdz”. Derīguma termiņš attiecas uz mēneša pēdējo dienu.
- Šīm zālēm nav nepieciešami īpaši uzglabāšanas apstākļi.
- Nelietojiet šīs zāles, ja pamanāt, ka iepakojums ir bojāts vai ir redzamas viltošanas pazīmes.
Neizmetiet zāles kanalizācijā vai sadzīves atkritumos. Jautājiet farmaceitam, kā izmest zāles, kuras vairs nelietojat. Tas palīdzēs aizsargāt vidi.
Ko Sutent satur
Sutent 12,5 mg cietās kapsulas
Aktīvā viela ir sunitinibs. Katra kapsula satur sunitiniba malātu, kas atbilst 12,5 mg sunitiniba.
Citas sastāvdaļas ir:
- Kapsulas saturs: mannīts (E421), nātrija kroskarmeloze, povidons (K-25) un magnija stearāts.
- Kapsulas apvalks: želatīns, sarkanais dzelzs oksīds (E172) un titāna dioksīds (E171).
- Tinte: šellaks, propilēnglikols, nātrija hidroksīds, povidons un titāna dioksīds (E171).
Sutent 25 mg cietās kapsulas
Aktīvā viela ir sunitinibs. Katra kapsula satur sunitiniba malātu, kas atbilst 25 mg sunitiniba.
Citas sastāvdaļas ir:
- Kapsulas saturs: mannīts (E421), nātrija kroskarmeloze, povidons (K-25) un magnija stearāts.
- Kapsulas apvalks: želatīns, titāna dioksīds (E171), dzeltenais dzelzs oksīds (E172), sarkanais dzelzs oksīds (E172), melnais dzelzs oksīds (E172).
- Tinte: šellaks, propilēnglikols, nātrija hidroksīds, povidons un titāna dioksīds (E171)
Sutent 37,5 mg cietās kapsulas
Aktīvā viela ir sunitinibs. Katra kapsula satur sunitiniba malātu, kas atbilst 37,5 mg sunitiniba.
Citas sastāvdaļas ir:
- Kapsulas saturs: mannīts (E421), nātrija kroskarmeloze, povidons (K-25) un magnija stearāts.
- Kapsulas apvalks: želatīns, titāna dioksīds (E171), dzeltenais dzelzs oksīds (E172).
- Tinte: šellaks, propilēnglikols, kālija hidroksīds, melnais dzelzs oksīds (E172).
Sutent 50 mg cietās kapsulas
Aktīvā viela ir sunitinibs. Katra kapsula satur sunitiniba malātu, kas atbilst 50 mg sunitiniba.
Citas sastāvdaļas ir:
- Kapsulas saturs: mannīts (E421), nātrija kroskarmeloze, povidons (K-25) un magnija stearāts.
- Kapsulas apvalks: želatīns, titāna dioksīds (E171), dzeltenais dzelzs oksīds (E172), sarkanais dzelzs oksīds (E172) un melnais dzelzs oksīds (E172).
- Tinte: šellaks, propilēnglikols, nātrija hidroksīds, povidons un titāna dioksīds (E171).
Sutent ārējais izskats un iepakojums
Sutent 12,5 mg ir pieejamas cietās želatīna kapsulās ar oranžu vāciņu un korpusu ar uzrakstu "Pfizer" ar baltu tinti uz vāciņa un "STN 12,5 mg" uz korpusa, kas satur krāsainas granulas.
Sutent 25 mg ir pieejamas cietās želatīna kapsulās ar karameļu vāciņu un oranžu korpusu, ar uzrakstu "Pfizer" ar baltu tinti un "STN 25 mg" uz korpusa, kas satur krāsainas granulas.
Sutent 37,5 mg ir pieejamas cietās želatīna kapsulās ar dzeltenu vāciņu un korpusu ar uzrakstu "Pfizer" ar melnu tinti uz vāciņa un "STN 37,5 mg" uz korpusa, kas satur krāsainas granulas.
Sutent 50 mg ir pieejamas cietās želatīna kapsulās ar karameļkrāsas vāciņu un korpusu ar uzrakstu "Pfizer" ar baltu tinti uz vāciņa un "STN 50 mg" uz korpusa, kas satur dzeltenīgi oranžas granulas. Tas ir pieejams pudelēs pa 30 kapsulām un perforētos blisteros ar vienreizēju devu, kas satur 28 x 1 kapsulas.
Visi iepakojuma lielumi tirgū var nebūt pieejami.
Avota lietošanas instrukcija: AIFA (Itālijas zāļu aģentūra). Saturs publicēts 2016. gada janvārī. Pašlaik pieejamā informācija var nebūt atjaunināta.
Lai piekļūtu visjaunākajai versijai, ieteicams piekļūt AIFA (Itālijas zāļu aģentūra) vietnei. Atruna un noderīga informācija.
01.0 ZĀĻU NOSAUKUMS
SUTENT 12,5 MG CIETAS KAPSULAS
02.0 KVALITATĪVAIS UN KVANTITATĪVAIS SASTĀVS
Katra kapsula satur sunitiniba malātu, kas atbilst 12,5 mg sunitiniba.
Pilnu palīgvielu sarakstu skatīt apakšpunktā 6.1.
03.0 ZĀĻU FORMA
Cieta kapsula.
Želatīna kapsulas ar oranžu vāciņu un korpusu, uz vāka ar baltu tinti apzīmētu "Pfizer", uz korpusa-"STN 12,5 mg" un satur dzeltenīgi oranžas granulas.
04.0 KLĪNISKĀ INFORMĀCIJA
04.1 Terapeitiskās indikācijas
Kuņģa -zarnu trakta stromas audzējs (GIST)
SUTENT ir indicēts neatgriezeniska un / vai metastātiska kuņģa -zarnu trakta stromas vēža (GIST) ārstēšanai pieaugušajiem pēc imatiniba terapijas neveiksmes rezistences vai nepanesības dēļ.
Metastātiska nieru šūnu karcinoma (MRCC)
SUTENT indicēts progresējošas / metastātiskas nieru šūnu karcinomas (MRCC) ārstēšanai pieaugušajiem.
Aizkuņģa dziedzera neiroendokrīnie audzēji (pNET)
SUTENT ir indicēts labi diferencētu, neatgriezenisku vai metastātisku aizkuņģa dziedzera neiroendokrīno audzēju (pNET) ārstēšanai progresējošas slimības gadījumā pieaugušajiem.
Pieredze ar SUTENT kā pirmās izvēles zālēm ir ierobežota (skatīt apakšpunktu 5.1).
04.2 Devas un lietošanas veids
Sunitiniba terapiju drīkst uzsākt ārsts, kuram ir pieredze pretvēža līdzekļu lietošanā.
Devas
GIST un MRCC gadījumā ieteicamā SUTENT deva ir 50 mg iekšķīgi vienu reizi dienā 4 nedēļas pēc kārtas, kam seko 2 nedēļu atpūta (4./2. Shēma), lai veiktu pilnu 6 nedēļu kursu.
PNET gadījumā ieteicamā SUTENT deva ir 37,5 mg, ko lieto iekšķīgi vienu reizi dienā bez plānotā atpūtas perioda.
Devas pielāgošana
Drošība un panesamība
GIST un MRCC gadījumā devu var mainīt ar 12,5 mg soli, pamatojoties uz pacienta individuālo drošību un panesamību. Dienas deva nedrīkst pārsniegt 75 mg, un to nedrīkst samazināt zem 25 mg.
Attiecībā uz pNET devu var pielāgot ar 12,5 mg soli, pamatojoties uz pacienta individuālo drošību un panesamību. Maksimālā deva, kas tika ievadīta 3. fāzes pNET pētījumā, bija 50 mg dienā.
Var būt nepieciešams pārtraukt dažu devu lietošanu, ņemot vērā pacienta drošību un panesamību.
CYP3A4 inhibitori / induktori
Jāizvairās no sunitiniba vienlaicīgas lietošanas ar spēcīgiem CYP3A4 induktoriem, piemēram, rifampicīnu (skatīt 4.4. Un 4.5. Apakšpunktu). Ja tas nav iespējams, var būt nepieciešams palielināt sunitiniba devu ar 12,5 mg soli (līdz 87,5 mg dienā GIST un MRCC vai 62,5 mg dienā pNET gadījumā), pamatojoties uz rūpīgu panesamības uzraudzību.
Jāizvairās no sunitiniba vienlaicīgas lietošanas ar spēcīgiem CYP3A4 inhibitoriem, piemēram, ketokonazolu (skatīt 4.4. Un 4.5. Apakšpunktu). Ja tas nav iespējams, var būt nepieciešams samazināt sunitiniba devu līdz minimālajai devai - 37,5 mg dienā GIST un MRCC gadījumā vai 25 mg dienā pNET gadījumā, pamatojoties uz rūpīgu panesamības uzraudzību.
Jāapsver iespēja izvēlēties alternatīvas vienlaicīgas zāles, kurām nav vai ir minimāla iespēja inducēt vai inhibēt CYP3A4.
Īpašas populācijas
Pediatriskā populācija
Sunitiniba drošība un efektivitāte pacientiem līdz 18 gadu vecumam nav noteikta.
Dati nav pieejami.
Nav indikāciju īpašai sunitiniba lietošanai bērniem no dzimšanas līdz 6 gadu vecumam neatgriezenisku un / vai metastātisku GIST ārstēšanā pēc imatiniba terapijas neveiksmes rezistences vai nepanesības dēļ. Nav indikāciju īpašai sunitiniba lietošanai pediatriskā populācijā MRCC ārstēšanā un labi diferencētu, nerezervējamu vai metastātisku pNET ārstēšanā slimības progresēšanas laikā.
Sunitiniba lietošana pediatriskā populācijā nav ieteicama.
Gados vecāki pacienti (≥ 65 gadus veci))
Aptuveni viena trešdaļa klīniskajos pētījumos iesaistīto pacientu, kuri saņēma sunitinibu, bija 65 gadus veci vai vecāki.Nav novērotas būtiskas atšķirības drošībā un efektivitātē starp jaunākiem un vecākiem cilvēkiem.
Aknu darbības traucējumi
Sākotnējās devas pielāgošana nav nepieciešama, ja sunitinibu lieto pacientiem ar viegliem vai vidēji smagiem aknu darbības traucējumiem (Child-Pugh A un B stadija). Sunitiniba lietošana cilvēkiem ar smagiem aknu darbības traucējumiem (C pakāpe pēc Child-Pugh) nav pētīta, tāpēc nav ieteicams to lietot pacientiem ar aknu darbības traucējumiem (skatīt 5.2. Apakšpunktu).
Nieru darbības traucējumi
Sākotnējā deva nav jāpielāgo, ja sunitinibu lieto pacientiem ar nieru darbības traucējumiem (vidēji smagi vai smagi) vai nieru slimību beigu stadijā (ESRD), kuriem tiek veikta hemodialīze. Turpmākā devas pielāgošana jāveic, ņemot vērā pacienta drošību un panesamību (skatīt 5.2. Apakšpunktu).
Lietošanas veids
SUTENT paredzēts iekšķīgai lietošanai. To var lietot kopā ar ēdienu vai bez tā.
Ja deva netiek lietota, papildu devu nedrīkst ievadīt. Nākamajā dienā pacientam jālieto ierastā deva.
04.3 Kontrindikācijas
Paaugstināta jutība pret aktīvo vielu vai jebkuru no 6.1. Apakšpunktā uzskaitītajām palīgvielām.
04.4 Īpaši brīdinājumi un piesardzība lietošanā
Jāizvairās no vienlaicīgas lietošanas ar spēcīgiem CYP3A4 induktoriem, jo tas var samazināt sunitiniba koncentrāciju plazmā (skatīt 4.2. Un 4.5. Apakšpunktu).
Jāizvairās no vienlaicīgas lietošanas ar spēcīgiem CYP3A4 inhibitoriem, jo tas var palielināt sunitiniba koncentrāciju plazmā (skatīt 4.2. Un 4.5. Apakšpunktu).
Ādas un audu bojājumi
Ādas krāsas maiņa, ko var izraisīt aktīvās vielas krāsa (dzeltena), ir ļoti bieži sastopama blakusparādība, kas rodas aptuveni 30% pacientu. Pacienti jābrīdina, ka ārstēšanas laikā ar sunitinibu tie var mainīt matu vai var rasties arī āda. Citi iespējamie dermatoloģiskie efekti var būt ādas sausums, sabiezējums vai plaisāšana, pūslīši vai neregulāri izsitumi uz plaukstām vai pēdām.
Iepriekš minētās reakcijas nebija kumulatīvas, tās parasti bija atgriezeniskas un parasti neizraisīja ārstēšanas pārtraukšanu. Ir ziņots par gangrenozas piodermijas gadījumiem, kas parasti ir atgriezeniski pēc zāļu lietošanas pārtraukšanas. Ir ziņots par nopietnām ādas reakcijām, tostarp multiformas eritēmas gadījumiem ( EM) un gadījumi, kas saistīti ar Stīvensa-Džonsona sindromu (SJS) un toksisku epidermas nekrolīzi (NET), daži no tiem ir letāli. sunitiniba lietošana jāpārtrauc. Ja tiek apstiprināta SJS vai NET diagnoze, ārstēšanu nevajadzētu atsākt. Dažos gadījumos, kad ir aizdomas par EM, pēc reakcijas izzušanas pacienti atkārtoti ievada sunitiniba terapiju ar mazāku panesamību; daži no šiem pacientiem arī saņēma vienlaicīga ārstēšana ar kortikosteroīdiem vai antiseptiķiem tamines.
Asiņošana un asiņošana, ko izraisa audzēji
Pēcreģistrācijas laikā ziņots par hemorāģiskiem notikumiem, dažos gadījumos ar letālu iznākumu, tai skaitā kuņģa-zarnu trakta, elpošanas, urīna un smadzeņu asiņošanu.
Asiņošanas epizodes novēroja 18% pacientu, kuri tika ārstēti ar sunitinibu, salīdzinot ar 17% pacientu placebo grupā GIST 3. fāzes pētījumā. Pacienti ar MRCC, kuri lietoja sunitinibu, nekad iepriekš nebija ārstēti ar sunitinibu, ziņoja par asiņošanu 39% gadījumu, salīdzinot ar 11% ar IFN-α ārstētiem pacientiem. Septiņpadsmit (4,5%) pacientiem, kuri lietoja sunitinibu, salīdzinot ar 5 (1,7%) pacientiem, kuri lietoja IFN-α, novēroja 3. vai augstākas pakāpes asiņošanas epizodes. Divdesmit seši procenti pacientu, kas saņēma sunitinibu citokīnu refrakcijas MRCC dēļ, ziņoja par asiņošanas epizodēm. Asiņošanas gadījumi, izņemot deguna asiņošanu, parādījās 21,7% pacientu, kuri tika ārstēti ar sunitinibu, salīdzinot ar 9,85% pacientu placebo grupā 3. fāzes pNET pētījumā. Šī notikuma regulārajā novērtēšanā jāiekļauj pilnīga asins analīze un fiziska pārbaude.
Deguna asiņošana bija visizplatītākā nevēlamā hemorāģiskā reakcija, par ko ziņots aptuveni pusei pacientu ar cietiem audzējiem, kuri ziņoja par asiņošanas gadījumiem.
Ir ziņots par audzēja asiņošanu, kas dažkārt ir saistīta ar audzēja nekrozi; daži no šiem asiņošanas gadījumiem bija letāli.
Klīniskajos pētījumos "audzēja asiņošana radās aptuveni 2% pacientu ar GIST. Šie notikumi var rasties pēkšņi un plaušu vēža gadījumā var izpausties kā" smaga un dzīvībai bīstama hemoptīze vai kā plaušu asiņošana..Klīniskajos pētījumos ir novēroti plaušu asiņošanas gadījumi, daži ar letālu iznākumu, un par tiem ziņots arī pēcreģistrācijas periodā pacientiem, kuri ārstēti ar sunitinibu MRCC, GIST un plaušu vēža dēļ. SUTENT nav apstiprināts lietošanai pacientiem ar plaušu vēzi.
Pacientus, kuri vienlaikus tiek ārstēti ar antikoagulantiem (piemēram, varfarīnu, acenokumarolu), var periodiski kontrolēt, veicot pilnu asins analīzi (trombocītu), koagulācijas faktorus (PT / INR) un veicot fizisku pārbaudi.
Kuņģa -zarnu trakta traucējumi
Caureja, slikta dūša / vemšana, sāpes vēderā, dispepsija un stomatīts / sāpes mutē bija visbiežāk ziņotās kuņģa -zarnu trakta blakusparādības; Ir ziņots arī par ezofagīta gadījumiem (skatīt apakšpunktu 4.8).
Kuņģa-zarnu trakta nevēlamo blakusparādību, kurām nepieciešama ārstēšana, atbalstoša aprūpe var ietvert zāles ar pretvemšanas, pretcaurejas vai antacīdu īpašībām.
Pacientiem ar intraabdominālu ļaundabīgu audzēju, kas ārstēti ar sunitinibu, ir radušās nopietnas, dažkārt letālas kuņģa-zarnu trakta komplikācijas, tai skaitā kuņģa-zarnu trakta perforācija. 3. fāzes GIST pētījumā letāla kuņģa -zarnu trakta asiņošana notika 0,98% pacientu, kuri tika ārstēti ar placebo.
Hipertensija
Hipertensija bija ļoti bieži sastopama blakusparādība, par ko ziņots klīniskajos pētījumos. Sunitiniba deva tika samazināta vai tā lietošana uz laiku tika pārtraukta aptuveni 2,7% pacientu, kuriem radās hipertensija. Nevienam no šiem pacientiem sunitiniba lietošana tika neatgriezeniski pārtraukta. Smaga hipertensija (> 200 mmHg sistoliskais vai 110 mmHg diastoliskais) novēroja 4,7% pacientu ar cietiem audzējiem. MRCC un iepriekš ārstētiem pacientiem par hipertensijas gadījumiem ziņoja 33, 9% pacientu, kuri lietoja sunitinibu, salīdzinot ar 3,6% pacientu, kuri lietoja IFN-α. smaga hipertensija radās 12% iepriekš neārstētu pacientu sunitiniba grupā un mazāk nekā 1% IFN-α grupas pacientu. 3. fāzes pNET pētījumā par hipertensiju ziņots 26,5% pacientu, kuri lietoja sunitinibu, salīdzinot ar 4,9% pacientu placebo grupā. Smagas hipertensijas epizodes pacientiem ar pNET radās 10% pacientu, kuri tika ārstēti ar sunitinibu un 3% pacientiem, kas ārstēti ar placebo.Pacienti jāpārbauda attiecībā uz hipertensiju un atbilstoši jākontrolē. Pacientiem ar smagu nekontrolētu hipertensiju, ārstējot ar zālēm, ieteicams īslaicīgi pārtraukt ārstēšanu. Ārstēšanu var atsākt, ja hipertensija ir pietiekami kontrolēta.
Hematoloģiski traucējumi
Absolūts 3. un 4. pakāpes neitrofilo leikocītu skaita samazinājums tika novērots attiecīgi 10% un 1,7% pacientu, kas iesaistīti 3. fāzes GIST pētījumā, un 16% un 1,6% pacientu, kas tika iekļauti pētījumā. 13% un 2,4% pacientu, kas tika iekļauti 3. fāzes pNET pētījumā. Par 3. un 4. pakāpes trombocītu skaita samazināšanos ziņots attiecīgi 3,7% un 0,4% pacientu. Pacienti, kas iesaistīti 3. fāzes GIST pētījumā, 8,2% un 1,1% pacientu, kas iekļauti MRCC 3. fāzes pētījumā, un 3,7% un 1,2% pacientu, kas iekļauti 3. fāzes pētījumā par pNET.
Iepriekš minētie notikumi nebija kumulatīvi, parasti bija atgriezeniski un parasti neizraisīja ārstēšanas pārtraukšanu. Neviens no šiem notikumiem 3. fāzes pētījumos nebija letāls, bet pēcreģistrācijas periodā ziņots par retiem hematoloģiskiem notikumiem. ieskaitot asiņošanu, kas saistīta ar trombocitopēniju un neitropēniskām infekcijām.
Ir novērota anēmija gan sunitiniba terapijas agrīnajā, gan vēlīnā stadijā; ziņots par 3. un 4. pakāpi.
Pacientiem, kuri saņem sunitinibu, katra ārstēšanas cikla sākumā jāveic pilnīga asins analīze.
Sirds patoloģijas
Pacientiem, kas ārstēti ar sunitinibu, ziņots par sirds un asinsvadu sistēmas traucējumiem, dažos gadījumos letālos, tostarp sirds mazspēju, kardiomiopātiju, miokarda išēmiju un miokarda infarktu. Šie dati liecina, ka sunitinibs palielina kardiomiopātijas risku. Ārstētiem pacientiem nav identificēti citi īpaši sunitiniba izraisītas kardiomiopātijas riska faktori, izņemot zāļu īpašo iedarbību. Sunitinibs jālieto piesardzīgi pacientiem, kuriem ir šo notikumu risks vai kuriem ir bijuši šādi notikumi.
Klīniskajos pētījumos kreisā kambara izsviedes frakcijas (LVEF) samazināšanās ≥ 20% un zem normas apakšējās robežas radās aptuveni 2% ar sunitinibu ārstētu GIST pacientu, 4% pacientu ar MRCC, kuri nebija izturīgi pret citokīniem, kuri tika ārstēti ar sunitinibu, un 2% % no placebo ārstētiem GIST pacientiem. Šķiet, ka šis LVEF samazinājums nav progresīvs un bieži vien uzlabojas, turpinot ārstēšanu. Pētījumā, kas tika veikts pacientiem ar MRCC un nekad iepriekš nebija ārstēts, 27% ar sunitinibu ārstēto pacientu un 15% ar IFN-α ārstēto pacientu LVEF vērtība bija zemāka par normas apakšējo robežu. Diviem pacientiem (
Pacientiem ar GIST par "sirds mazspējas", "sastrēguma sirds mazspējas" vai "kreisā kambara mazspējas" epizodēm ziņoja 1,2% ar sunitinibu ārstēto pacientu un 1% pacientu, kuri tika ārstēti ar placebo. pēc GIST (n = 312) ar ārstēšanu saistītas letālas sirds reakcijas radās 1% pacientu abās pētījuma grupās (sunitiniba un placebo grupā). Otrās fāzes pētījumā, kurā piedalījās pacienti ar citokīniem necaurlaidīgu MRCC, 0,9 % pacientu ziņoja par ar ārstēšanu saistītu letālu miokarda infarktu, bet 3. fāzes pētījumā ar pacientiem ar MRCC un nekad iepriekš neārstētu-0,6 % pacientu IFN-α grupā un 0% pacientu sunitiniba grupā ziņoja par letāliem sirdsdarbības gadījumiem. 3. fāzes pNET pētījumā vienam pacientam (1%), kas lietoja sunitinibu, bija ar ārstēšanu saistīta letāla sirds mazspēja. Iespējamā korelācija starp tirozīnkināzes receptoru (RTK) inhibīciju un sirds funkciju nav skaidra.
Pacienti, kuriem 12 mēnešu laikā pirms sunitiniba ievadīšanas ir bijuši sirdsdarbības traucējumi, piemēram, miokarda infarkts (ieskaitot smagu / nestabilu stenokardiju), koronāro / perifēro šuntēšanas operācija, simptomātiska sirds mazspēja, cerebrovaskulāri traucējumi vai pārejoša išēmiska lēkme vai plaušu embolija, tika izslēgti no klīniskajiem pētījumiem. izmēģinājumi ar sunitinibu. Nav zināms, vai pacientiem ar šādiem vienlaicīgiem stāvokļiem var būt paaugstināts ar zālēm saistītas kreisā kambara disfunkcijas risks.
Ir rūpīgi jāuzrauga sastrēguma sirds mazspējas klīniskās pazīmes un simptomi, īpaši pacientiem ar sirds riska faktoriem un / vai ar koronāro artēriju slimību.
Ārstiem ieteicams šo risku salīdzināt ar iespējamo zāļu ieguvumu. Ārstēšanas ar sunitinibu laikā šie pacienti rūpīgi jānovēro, vai nav konstatētas sastrēguma sirds mazspējas klīniskās pazīmes un simptomi. Ja pacients tiek ārstēts ar sunitinibu, jāņem vērā arī sākotnējais un periodiskais kreisā kambara izsviedes frakcijas novērtējums. Pacientiem, kuriem nav sirds riska faktoru, sākotnēji jāapsver ventrikulārās izsviedes frakcijas novērtējums.
Ja ir CHF klīniskās izpausmes, sunitiniba terapiju ieteicams pārtraukt. Pacientiem, kuriem nav klīnisku pierādījumu par sastrēguma sirds mazspēju, bet sunitiniba lietošana jāpārtrauc un / vai jāsamazina deva, bet izsviedes frakcija samazinās no 20% līdz 50% no bāzes līnijas.
QT intervāla pagarināšana
Preklīnisko pētījumu dati (in vitro Un in vivo), kuras tika veiktas ar lielākām devām nekā ieteicamās cilvēkiem, norāda, ka sunitinibs var kavēt sirds repolarizācijas procesus (piemēram, pagarināt QT intervālu).
QTc intervāla palielināšanās līdz vairāk nekā 500 ms radās ar 0,5% ātrumu, un izmaiņas no sākotnējā stāvokļa vairāk nekā 60 ms radās 1,1% no 450 pacientiem ar cietiem audzējiem; abi šie parametri tiek atzīti par potenciāli nozīmīgām variācijām. Koncentrācijās, kas aptuveni divas reizes pārsniedz terapeitisko koncentrāciju, tika konstatēts, ka sunitinibs pagarina QTcF intervālu (koriģēts pēc Frederikas formulas).
QT intervāla pagarināšanos pētīja pētījumā, kurā piedalījās 24 pacienti vecumā no 20 līdz 87 gadiem ar progresējošiem ļaundabīgiem audzējiem. Šī pētījuma rezultāti parādīja, ka sunitinibs ietekmēja QTc intervālu (definēts kā ar placebo koriģētas vidējās izmaiņas> 10 ms, ar augšējā robeža 90% TI> 15 ms) terapeitiskā koncentrācijā (3. diena), izmantojot 24 stundu sākotnējās korekcijas metodi, un koncentrācijās, kas pārsniedz terapeitisko (9. diena), izmantojot abas korekcijas metodes sākotnējā stāvoklī. Neviens pacients nav ziņojis par QTc vērtību> 500 ms. Lai gan ietekme uz QTcF intervālu tika novērota 3. dienā 24 stundas pēc devas (t.i., ar paredzamo terapeitisko koncentrāciju plazmā pēc ieteicamās sākuma devas 50 mg), izmantojot 24 stundu sākotnējās korekcijas metodi, šī konstatējuma klīniskā nozīme nav skaidra. .
Izmantojot visaptverošus sērijveida EKG novērtējumus, kas atbilst terapeitiskajai iedarbībai vai iedarbībai, kas pārsniedz terapeitisko, nevienam no pacientiem novērtējamajās vai ITT populācijās netika novērots "smaga" QTc intervāla pagarinājums (tāpēc vienāds vai lielāks par
CTCAE 3.0 versijas 3. pakāpe).
Terapeitiskās koncentrācijas plazmā vidējās maksimālās QTcF intervāla izmaiņas (koriģētas pēc Frederikas formulas) salīdzinājumā ar sākotnējo rādītāju bija 9,6 ms (90% TI 15,1 ms). Terapeitiskās koncentrācijās, kas atbilst aptuveni divreiz lielākai terapeitiskajai koncentrācijai, maksimālās QTcF intervāla izmaiņas salīdzinājumā ar sākotnējo bija 15,4 ms (90% TI 22,4 ms).
Moksifloksacīnam (400 mg), ko izmantoja kā pozitīvu kontroli, vidējās maksimālās QTcF intervāla izmaiņas salīdzinājumā ar sākotnējo rādītāju bija 5,6 ms. Neviens subjekts nepaziņoja par ietekmi uz QTc intervālu, kas būtu lielāks par 2. pakāpi (CTCAE versija 3.0).
QT intervāla pagarināšanās var palielināt sirds kambaru aritmiju, tai skaitā Torsade de Pointes, risku.
Sunitinibs piesardzīgi jālieto pacientiem, kuriem anamnēzē ir pagarināts QT intervāls, pacientiem, kas ārstēti ar antiaritmiskiem līdzekļiem vai ar zālēm, kas var pagarināt QT intervālu, vai pacientiem, kuriem jau ir bijusi atbilstoša sirds slimība, bradikardija vai anomālijas. Sunitiniba vienlaicīga lietošana ar spēcīgiem CYP3A4 inhibitoriem jāierobežo, jo iespējama sunitiniba koncentrācijas palielināšanās plazmā (skatīt 4.2. Un 4.5. Apakšpunktu).
Venozas trombembolijas gadījumi
Ar ārstēšanu saistītus vēnu trombembolijas gadījumus klīniskajos pētījumos, tostarp GIST un MRCC pētījumos, novēroja aptuveni 1,0% pacientu ar cietiem audzējiem, kuri tika ārstēti ar sunitinibu.
3. fāzes GIST pētījumā septiņiem pacientiem (3%), kuri saņēma sunitinibu, un nevienam pacientam placebo grupā nebija vēnu trombembolijas; pieciem no septiņiem pacientiem bija 3. pakāpes dziļo vēnu tromboze (DVT), bet diviem - 1. vai 2. pakāpes dziļo vēnu tromboze.Četri no šiem septiņiem pacientiem, kuri tika ārstēti ar GIST, tika pārtraukti pēc DVT novērošanas.
Trīspadsmit pacienti (3%), kuri tika ārstēti ar sunitinibu MRCC 3. fāzes pētījumā un nekad iepriekš nebija ārstēti, un četri pacienti (2%) no diviem citokīnu refraktīviem MRCC pētījumiem ziņoja par vēnu trombemboliskiem notikumiem. Deviņiem no šiem pacientiem bija vēnu trombembolijas gadījumi. " plaušu embolija, vienam ar 2. pakāpi un astoņiem ar 4. pakāpi. Astoņiem no šiem pacientiem bija DVT, vienam - 1. pakāpe, diviem - 2. pakāpe, četriem - 3. pakāpe un vienam - 4. pakāpe. Pacientam ar plaušu emboliju MRCC pētījumā , kas nav izturīgi pret citokīniem, deva tika pārtraukta.
No iepriekš neārstētiem MRCC pacientiem, kuri saņēma IFN-α, seši (2%) ziņoja par vēnu trombembolijas gadījumiem, viens pacients (plaušu embolija, visi 4. pakāpe).
3. fāzes pNET pētījumā par vēnu trombemboliju ziņots 1 (1,2%) sunitiniba grupas pacientam un 5 (6,1%) pacientiem placebo grupā. Divi no šiem placebo ārstētajiem ziņoja par DVT, viens 2. un 3. pakāpe.
GIST, MRCC un pNET pamatpētījumos netika novēroti letāli gadījumi. Pēcreģistrācijas periodā novēroti gadījumi ar letālu iznākumu (skatīt elpošanas traucējumus un 4.8. Apakšpunktu).
Arteriālās trombembolijas gadījumi
Pacientiem, kuri tika ārstēti ar sunitinibu, ziņots par arteriālas trombembolijas gadījumiem (ATE), dažkārt letālu. Visbiežāk sastopamie notikumi bija smadzeņu asinsvadu negadījums, pārejošs išēmisks uzbrukums un smadzeņu išēmija. Riska faktori, kas saistīti ar artēriju trombembolijas gadījumiem, papildus jau esošam ļaundabīgam audzējam un 65 gadu vecumam vai vecākiem, ietvēra hipertensiju, cukura diabētu un iepriekšēju trombembolisku notikumu.
Elpošanas traucējumi
Pacienti, kuriem pēdējo 12 mēnešu laikā bija plaušu embolija, tika izslēgti no sunitiniba klīniskajiem pētījumiem.
Pacientiem, kuri saņēma sunitinibu galvenajos 3. fāzes pētījumos, plaušu notikumi (aizdusa, pleiras izsvīdums, plaušu embolija vai plaušu tūska) tika novēroti aptuveni 17,8% pacientu ar GIST, aptuveni 26,7% pacientu ar MRCC un 12% pacientu pacientiem ar pNET.
Aptuveni 22,2% pacientu ar cietiem audzējiem, ieskaitot GIST un MRCC, klīniskajos pētījumos ārstēja ar sunitinibu, ziņoja par plaušu parādībām.
Plaušu embolijas gadījumi tika novēroti aptuveni 3,1% pacientu ar GIST un aptuveni 1,2% pacientu ar MRCC, kuri tika ārstēti ar sunitinibu 3. fāzes pētījumos (skatīt apakšpunktu 4.4. - Venozas trombembolijas gadījumi). PNET pacientiem plaušu embolijas gadījumi netika novēroti. ārstēti ar sunitinibu 3. fāzes pētījumā .. Pēcreģistrācijas periodā ir novēroti reti gadījumi ar letālu iznākumu (skatīt 4.8. apakšpunktu).
Vairogdziedzera disfunkcija
Visiem pacientiem ieteicams novērtēt vairogdziedzera darbību, izmērot sākotnējās laboratorijas vērtības. Pacienti ar hipotireozi vai hipertireozi pirms sunitiniba terapijas uzsākšanas jāārstē saskaņā ar standarta klīnisko praksi. Ārstēšanas laikā ar sunitinibu ik pēc 3 mēnešiem jāveic regulāra vairogdziedzera funkcijas pārbaude. Turklāt visi pacienti ārstēšanas laikā rūpīgi jānovēro, vai nav iespējamu vairogdziedzera disfunkcijas pazīmju un simptomu, un pacientiem, kuriem rodas jebkādas pazīmes un / vai simptomi, kas liecina par vairogdziedzera darbības traucējumiem, jāveic laboratoriska vairogdziedzera funkcijas pārbaude, kā klīniski paredzēts. Pacienti, kuriem attīstās vairogdziedzera darbības traucējumi, jāārstē saskaņā ar standarta klīnisko praksi.
Ir ziņots, ka sunitiniba terapijas sākumā vai beigās rodas hipotireoze.
Divos MRCC pētījumos, kas tika veikti ar citokīnu refraktāriem pacientiem, tika ziņots par hipotireozi kā blakusparādību 7 pacientiem (4%), kuri saņēma sunitinibu; 61 pacientam (16%), kas saņēma sunitinibu, un trim pacientiem (
Turklāt tika ziņots par TSH līmeņa paaugstināšanos 4 pacientiem (2%), kuri tika ārstēti ar citokīniem necaurlaidīgu MRCC. Kopumā 7% MRCC populācijas ziņoja par klīniskiem vai laboratoriskiem pierādījumiem par ar ārstēšanu saistītu hipotireozi. Iegūto hipotireozi novēroja 6,2% pacientu GIST pētījumā, kuri lietoja sunitinibu, salīdzinot ar 1% placebo grupā. 3. fāzes pNET pētījumā par hipotireozi ziņots 6 pacientiem (7,2%), kuri saņēma sunitinibu, un vienam pacientam (1,2%), kas saņēma placebo.
Vairogdziedzera funkcija tika perspektīvi uzraudzīta divos pētījumos ar krūts vēža slimniekiem; SUTENT nav apstiprināts krūts vēža ārstēšanai. Vienā pētījumā par hipotireozi ziņots 15 subjektiem (13,6%), kuri tika ārstēti ar sunitinibu, un 3 subjektiem (2,9%), kuri tika ārstēti ar standarta terapiju. Paaugstināts TSH līmenis tika ziņots 1 pacientam. (0,9%), kuri tika ārstēti ar sunitinibu, un nevienam subjektam ārstē ar standarta terapiju. Par hipertireozi netika ziņots nevienam subjektam, kas tika ārstēts ar sunitinibu, un tika ziņots 1 pacientam (1,0%), kurš saņēma standarta terapiju. Citā pētījumā hipotireoze tika ziņota kopumā 31 subjektam (13%), kuri tika ārstēti ar sunitinibu un 2 Pacientiem (0,8%), kuri tika ārstēti ar kapecitabīnu. Paaugstināts TSH līmenis tika ziņots 12 subjektiem (5,0%), kuri tika ārstēti ar sunitinibu, un nevienam, kas netika ārstēts ar kapecitabīnu. Par hipertireozi ziņots 4 subjektiem (1,7%), kuri tika ārstēti ar sunitinibu, un nevienam, kas netika ārstēts ar kapecitabīnu. Pazemināts TSH līmenis tika ziņots 3 subjektiem (1,3%), kuri tika ārstēti ar sunitinibu, un nevienam subjektam, kuri ārstēti ar kapecitabīnu. 2 pacienti (0,8%), kas ārstēti ar sunitinibu, un 1 subjekts (0,4%), kas ārstēti ar kapecitabīnu. T3 pieaugums tika ziņots 1 subjektam (0,8%), kurš tika ārstēts ar sunitinibu, un nevienam pacientam, kurš netika ārstēts ar kapecitabīnu.
Klīniskajos pētījumos un zāļu tirdzniecības fāzē retāk ziņots par hipertireozes gadījumiem, dažiem seko hipotireoze un tireoīdīta gadījumi.
Pankreatīts
Pacientiem ar vairākiem cietiem audzējiem, kuri saņēma sunitinibu, tika novērota seruma lipāzes un amilāzes aktivitātes palielināšanās. Pacientiem ar dažāda veida cietiem audzējiem seruma lipāzes aktivitātes palielināšanās bija pārejoša un parasti nebija saistīta ar pankreatīta pazīmēm un simptomiem.
Pankreatīts tika novērots retāk (
Ir ziņots par nopietnu aizkuņģa dziedzera traucējumu gadījumiem, daži ar letālu iznākumu.
Ja rodas pankreatīta simptomi, sunitiniba terapija jāpārtrauc un jānodrošina pacientiem atbilstoša atbalstoša aprūpe. 3. fāzes pNET pētījumā netika ziņots par ar ārstēšanu saistītu pankreatītu.
Hepatotoksicitāte
Pacientiem, kuri tika ārstēti ar sunitinibu, ir novērota hepatotoksicitāte. Aknu darbības traucējumi (alanīna transamināzes [ALAT], aspartātransamināzes [ASAT], bilirubīna līmenis) ir novēroti aknu mazspējas gadījumi, dažkārt ar letālu iznākumu, aknu mazspējas pazīmes vai simptomi, ārstēšana ar sunitinibu jāpārtrauc un pacientiem jānodrošina atbilstoša atbalstoša medicīniskā aprūpe.
Aknu un žultsceļu darbības traucējumi
Ārstēšana ar sunitinibu var būt saistīta ar holecistītu, ieskaitot alītisko holecistītu un enftemātisko holecistītu. Pivotālos klīniskos pētījumos holecistīta sastopamība bija 0,5%.
Ir ziņots par holecistīta pēcreģistrācijas gadījumiem.
Nieru funkcija
Ir ziņots par nieru darbības traucējumiem, nieru mazspēju un / vai akūtu nieru mazspēju, dažos gadījumos ar letālu iznākumu.
Riska faktori, kas saistīti ar nieru darbības traucējumiem / mazspēju pacientiem, kuri saņēma sunitinibu, papildus jau esošai nieru šūnu karcinomai ietvēra: vecums, cukura diabēts, esoši nieru darbības traucējumi, sirds mazspēja, hipertensija, sepse, dehidratācija / hipovolēmija un rabdomiolīze.
Sunitiniba terapijas turpināšanas drošība pacientiem ar vidēji smagu vai smagu proteīnūriju nav sistemātiski novērtēta.
Ir ziņots par proteīnūrijas gadījumiem un retiem nefrotiskā sindroma gadījumiem. Ieteicama sākotnējā urīna analīze, un pacienti jāuzrauga, vai nav iespējama proteīnūrijas attīstība vai pasliktināšanās.
Pacientiem ar nefrozes sindromu sunitiniba terapija jāpārtrauc.
Fistulas
Ja veidojas fistula, ārstēšana ar sunitinibu jāpārtrauc. Ir ierobežota informācija par ilgstošu sunitiniba terapiju pacientiem ar fistulām.
Brūču dzīšanas procesa traucējumi
Sunitiniba terapijas laikā ziņots par brūču dzīšanas traucējumiem. Nav veikti oficiāli klīniskie pētījumi par sunitiniba ietekmi uz brūču dzīšanu. Piesardzības nolūkos ieteicams uz laiku pārtraukt sunitiniba terapiju pacientiem, kuriem tiek veikta liela operācija. Klīniskā pieredze par laiku, kas nepieciešams, lai atsāktu. Terapija pēc lielas operācijas ir ierobežota. Tādēļ lēmums par sunitiniba terapijas atsākšanu pēc lielas operācijas jāpamato ar klīnisko spriedumu par atveseļošanos pēc operācijas.
Apakšžokļa / augšžokļa osteonekroze
Pacientiem, kas ārstēti ar SUTENT, ziņots par žokļa osteonekrozes gadījumiem. Vairumā gadījumu, par kuriem ziņots, pacienti iepriekš vai vienlaikus tika ārstēti ar intravenoziem bisfosfonātiem, ar kuriem tika identificēts žokļa osteonekrozes risks. Tādēļ jāievēro piesardzība, ja SUTENT un intravenozus bisfosfonātus lieto vienlaikus vai kombinācijā.
Invazīvās zobu intervences ir identificētas arī kā riska faktors. Pirms ārstēšanas ar SUTENT jāapsver zobu pārbaude un atbilstoša profilaktiska zobu aprūpe. Ja iespējams, jāizvairās no invazīvas zobu iejaukšanās pacientiem, kuri iepriekš lietojuši vai lieto intravenozus bisfosfonātus (skatīt 4.8. Apakšpunktu).
Paaugstināta jutība / angioneirotiskā tūska
Ja rodas paaugstinātas jutības reakcijas izraisīta tūska, sunitiniba terapija jāpārtrauc un jāsniedz standarta medicīniskā ārstēšana.
Nervu sistēmas traucējumi
Garšas traucējumi
Klīnisko pētījumu laikā tika ziņots par disgeiziju aptuveni 28% pacientu, kuri saņēma sunitinibu.
Krampji
Klīniskajos pētījumos ar sunitinibu un pēcreģistrācijas periodā tika novēroti krampju gadījumi pacientiem ar vai bez radioloģiskiem smadzeņu metastāžu pierādījumiem. Turklāt ir saņemts ierobežots skaits ziņojumu (galvassāpes, modrības samazināšanās, garīgās funkcijas traucējumi un redzes zudums, ieskaitot garozas aklumu, jākontrolē ar medicīnisku ārstēšanu, kas ietver hipertensijas kontroli. Ieteicams. ja notikums atrisinās, ārstēšanu var atsākt pēc ārstējošā ārsta ieskatiem.
Audzēja līzes sindroms (TLS)
Klīniskajos pētījumos reti tika novēroti audzēja sabrukšanas sindroma (TLS) gadījumi, dažos gadījumos ar letālu iznākumu, un par tiem ziņots pēcreģistrācijas periodā ar sunitinibu ārstētiem pacientiem. TLS riska faktori ir augsts audzēja slogs, jau esoša hroniska nieru mazspēja, oligūrija, dehidratācija, hipotensija un skābs urīns. Šie pacienti rūpīgi jāuzrauga un jāārstē atbilstoši klīniskajām indikācijām; šiem pacientiem jāapsver profilaktiskā hidratācija.
Infekcijas
Ir ziņots par nopietnu infekciju gadījumiem ar vai bez neitropēnijas, ieskaitot dažus gadījumus ar letālu iznākumu.
Infekcijas, ko visbiežāk novēro, ārstējot sunitinibu, ir vēža slimniekam raksturīgas infekcijas, piemēram, elpceļu, urīnceļu, ādas un sepse.
Ir ziņots par retiem, dažreiz letāliem, nekrotizējoša fascīta gadījumiem, tai skaitā starpenē. Pacientiem, kuriem attīstās nekrotizējošs fascīts, sunitiniba terapija jāpārtrauc un nekavējoties jāsāk atbilstoša ārstēšana.
Hipoglikēmija
Ārstēšanas ar sunitinibu laikā ziņots par glikozes līmeņa pazemināšanos asinīs, daži no tiem ir klīniski simptomātiski un apziņas zuduma dēļ nepieciešama hospitalizācija. Simptomātiskas hipoglikēmijas gadījumā sunitiniba terapija uz laiku jāpārtrauc. Pacientiem ar cukura diabētu regulāri jāpārbauda glikozes līmenis asinīs, lai novērtētu, vai cukura diabēta zāļu deva ir jāpielāgo, lai samazinātu hipoglikēmijas risku.
04.5 Mijiedarbība ar citām zālēm un citi mijiedarbības veidi
Mijiedarbības pētījumi veikti tikai pieaugušajiem.
Zāles, kas var palielināt sunitiniba koncentrācija plazmā
Veseliem brīvprātīgajiem vienreizējas sunitiniba devas lietošana kopā ar spēcīgu CYP3A4 inhibitoru ketokonazolu izraisīja kombinētā [sunitiniba + primārā metabolīta] Cmax un AUC0-∞ palielināšanos attiecīgi par 49% un 51%.
Sunitiniba lietošana kopā ar spēcīgiem CYP3A4 inhibitoriem (piemēram, ritonavīru) , itrakonazols, eritromicīns, klaritromicīns, greipfrūtu sula) var palielināt sunitiniba koncentrāciju.
Tāpēc jāizvairās no kombinācijas ar CYP3A4 inhibitoriem vai jāapsver alternatīvas zāles, kurām nav vai ir minimāla iespēja inhibēt CYP3A4.
Ja tas nav iespējams, SUTENT devu, iespējams, vajadzēs samazināt līdz vismaz 37,5 mg dienā GIST un MRCC gadījumā vai līdz 25 mg dienā pNET gadījumā, pamatojoties uz rūpīgu panesamības uzraudzību (skatīt 4.2. Apakšpunktu).
Zāles, kas var samazināt sunitiniba koncentrācija plazmā
Veseliem brīvprātīgajiem, vienlaicīgi lietojot vienu sunitiniba un CYP3A4 inducētāja rifampicīna devu, kombinētais [sunitiniba + primārā metabolīta] Cmax un AUC0-∞ samazinājās attiecīgi par 23% un 46%.
Sunitiniba lietošana kopā ar spēcīgiem CYP3A4 induktoriem (piemēram, deksametazonu, fenitoīnu, karbamazepīnu, rifampicīnu, fenobarbitālu vai augu izcelsmes preparātiem, kas satur asinszāli /Hypericum perforatum) var samazināt sunitiniba koncentrāciju. Tāpēc jāizvairās no saistības ar CYP3A4 induktoriem vai jāapsver alternatīvas zāles, kurām nav vai ir minimāla iespēja inducēt CYP3A4. Ja tas nav iespējams, SUTENT devu var palielināt ar soli 12, 5 mg (līdz līdz 87,5 mg dienā GIST un MRCC vai 62,5 mg dienā pNET gadījumā), pamatojoties uz rūpīgu panesamības uzraudzību (skatīt 4.2. apakšpunktu).
04.6 Grūtniecība un zīdīšana
Grūtniecība
Nav veikti pētījumi ar grūtniecēm, kuras saņem sunitinibu. Pētījumi ar dzīvniekiem parādīja reproduktīvo toksicitāti, ieskaitot augļa malformācijas (skatīt 5.3. Apakšpunktu).
SUTENT nedrīkst lietot grūtniecības laikā vai sievietēm, kuras nelieto efektīvu kontracepciju, ja vien potenciālais ieguvums neattaisno iespējamo risku auglim. Ja SUTENT lieto grūtniecības laikā vai ja pacientam iestājas grūtniecība ārstēšanas laikā ar SUTENT, pacients jāinformē par iespējamo risku auglim.
Sievietēm reproduktīvā vecumā jāiesaka lietot efektīvus kontracepcijas līdzekļus un izvairīties no grūtniecības, ārstējoties ar SUTENT.
Barošanas laiks
Sunitinibs un / vai tā metabolīti izdalās žurku pienā. Nav zināms, vai sunitinibs vai tā galvenais aktīvais metabolīts izdalās mātes pienā. Tā kā aktīvās vielas parasti izdalās mātes pienā un ņemot vērā iespējamās nopietnās nevēlamās blakusparādības zīdainim, ārstēšanas laikā ar SUTENT sievietes nedrīkst barot bērnu ar krūti.
Auglība
Preklīniskie dati liecina, ka ārstēšana ar sunitinibu var pasliktināt vīriešu un sieviešu auglību (skatīt apakšpunktu 5.3).
04.7 Ietekme uz spēju vadīt transportlīdzekļus un apkalpot mehānismus
Nav veikti pētījumi par ietekmi uz spēju vadīt transportlīdzekļus un apkalpot mehānismus.Pacienti jāinformē par iespējamu reiboni, kas rodas ārstēšanas laikā ar sunitinibu.
04.8 Nevēlamās blakusparādības
Drošības profila kopsavilkums
Visnopietnākās ar sunitinibu saistītās blakusparādības, kas dažkārt ir letālas, ir nieru mazspēja, sirds mazspēja, plaušu embolija, kuņģa-zarnu trakta perforācija un asiņošana (piemēram, elpošanas, kuņģa-zarnu trakta, ar audzēju saistītas, urīna un smadzeņu asiņošanas).
Visizplatītākās jebkādas pakāpes blakusparādības (par kurām ziņoja pacienti galvenajos RCC, GIST un pNET klīniskajos pētījumos) bija apetītes samazināšanās, garšas traucējumi, hipertensija, nogurums, kuņģa -zarnu trakta traucējumi (piemēram, caureja, slikta dūša, stomatīts, dispepsija un vemšana), krāsas maiņa. ādas un plaukstas-plantāra eritrodizestēzijas sindroms. Šie simptomi var mazināties, turpinot ārstēšanu. Ārstēšanas laikā var rasties hipotireoze.Hematoloģiski traucējumi (piemēram, neitropēnija, trombocitopēnija un anēmija) bija vienas no biežākajām zāļu blakusparādībām.
Nāvējošas blakusparādības, kas nav uzskaitītas 4.4. apakšpunktā un, iespējams, saistīts ar sunitinibu, ietver vairāku orgānu mazspēju, izplatītu intravaskulāru koagulāciju, asiņošanu vēderplēvē, virsnieru mazspēju, pneimotoraksu, šoku un pēkšņu nāvi.
Blakusparādību tabula
Blakusparādības, par kurām ziņojuši pacienti ar GIST, MRCC un pNET apkopotā datu kopā ar 7115 pacientiem, ir uzskaitītas zemāk un iedalītas pēc orgānu sistēmas, biežuma un smaguma pakāpes (NCI-CTCAE). Ir ziņots arī par pēcreģistrācijas blakusparādībām, kas konstatētas klīniskajos pētījumos. Katrā sastopamības biežumā nevēlamās blakusparādības ir uzskaitītas to smaguma pakāpes samazinājuma secībā.
Biežums ir definēts šādi: ļoti bieži (≥1 / 10), bieži (≥1 / 100a
1. tabula. Klīniskajos pētījumos ziņotās blakusparādības
Ir sagrupēti šādi termini:
a nazofaringīts un mutes herpes
b Bronhīts, apakšējo elpceļu infekcija, pneimonija un elpceļu infekcija
c Abscess, ekstremitāšu abscess, anālais abscess, smaganu abscess, aknu abscess, aizkuņģa dziedzera abscess, starpenes abscess, perirectal abscess, taisnās zarnas abscess, zemādas abscess un zobu abscess
d Barības vada kandidoze un mutes kandidoze
un celulīts un ādas infekcija
f Sepsis un septisks šoks
g Vēdera abscess, vēdera sepsis, divertikulīts un osteomielīts
h Samazināta ēstgriba un anoreksija
i Disgeizija, ageūzija un garšas izmaiņas
j Akūts koronārais sindroms, stenokardija, nestabila stenokardija, koronāro artēriju oklūzija, miokarda išēmija
k Izstumšanas frakcijas samazināšana / anomālija
l Akūts miokarda infarkts, miokarda infarkts, kluss miokarda infarkts
m Sāpes rīklē un rīklē
n Stomatīts un aftozs stomatīts
o Sāpes vēderā, sāpes vēdera lejasdaļā un augšdaļā
p Kuņģa -zarnu trakta un zarnu perforācija
q holecistīts un alitiāzes holecistīts
r dzeltena āda, ādas krāsas izmaiņas un pigmentācijas traucējumi
s Psoriāzes formas dermatīts, eksfoliatīvi izsitumi, izsitumi, eritematozi izsitumi, folikulāri izsitumi, ģeneralizēti izsitumi, makulas izsitumi, makulopapulāri izsitumi, papulāri izsitumi un nieze
t Ādas reakcija un ādas patoloģija
u Nagu patoloģija un nagu krāsas maiņa
v Nogurums un astēnija
w Sejas tūska, tūska un perifēra tūska
x Amilāze un palielināta amilāze
* Ietver letālus notikumus
Konstatēto nevēlamo blakusparādību apraksts
Infekcijas un invāzijas: Ir ziņots par nopietnu infekciju gadījumiem (ar vai bez neitropēnijas), ieskaitot gadījumus ar letālu iznākumu. Ir ziņots par nekrotizējoša, dažkārt letāla fascīta gadījumiem, kas var ietekmēt arī starpenes zonu (skatīt arī apakšpunktu 4.4).
Asins un limfātiskās sistēmas traucējumi: Ir ziņots par trombotiskas mikroangiopātijas gadījumiem. Šādos gadījumos ieteicams uz laiku apturēt SUTENT lietošanu; pēc šo notikumu izzušanas ārstēšanu var atsākt pēc ārstējošā ārsta ieskatiem.
Imūnās sistēmas traucējumi: Ir ziņots par paaugstinātas jutības reakcijām, ieskaitot angioneirotisko tūsku.
Nervu sistēmas traucējumi: Ir ziņots par dažiem gadījumiem, dažos gadījumos ar letālu iznākumu, pacientiem ar krampjiem un radioloģiskiem pierādījumiem par atgriezenisku aizmugurējās leikoencefalopātijas sindromu (RPLS) (skatīt arī apakšpunktu 4.4).
Vielmaiņas un uztura traucējumi: Pacientiem ar pNET ziņots par lielāku hipoglikēmijas gadījumu skaitu nekā pacientiem ar MRCC un GIST. Tomēr daudzas klīniskajos pētījumos novērotās blakusparādības netika uzskatītas par saistītām ar pētījuma ārstēšanu.
Aknu un / vai žultsceļu traucējumi: Ir ziņots par aknu darbības traucējumiem, un tie var ietvert aknu darbības testu novirzes, hepatītu vai aknu mazspēju.
Ādas un zemādas audu bojājumi: Ir ziņots par gangrenozās piodermijas gadījumiem, kas parasti ir atgriezeniski pēc ārstēšanas pārtraukšanas (skatīt arī apakšpunktu 4.4).
Skeleta -muskuļu un saistaudu sistēmas bojājumi: Ir ziņots par miopātijas un / vai rabdomiolīzes gadījumiem, daži no tiem ir saistīti ar akūtu nieru mazspēju. Pacienti, kuriem ir muskuļu toksicitātes pazīmes vai simptomi, jāārstē saskaņā ar standarta medicīnas praksi.
Ir ziņots par fistulas veidošanās gadījumiem, kas dažkārt ir saistīti ar audzēja nekrozi un regresiju, dažos gadījumos ar letālu iznākumu.
Pacientiem, kas ārstēti ar SUTENT, ziņots par žokļa osteonekrozes gadījumiem, daudzi no tiem radās pacientiem, kuriem bija atzīti žokļa osteonekrozes riska faktori, īpaši intravenozu bifosfonātu iedarbība un / vai zobu slimības, kurām nepieciešama invazīva zobu iejaukšanās (sk. arī 4.4. apakšpunktu).
Ziņošana par iespējamām blakusparādībām
Ir svarīgi ziņot par iespējamām blakusparādībām, kas radušās pēc zāļu reģistrācijas, jo tas ļauj nepārtraukti uzraudzīt zāļu ieguvuma un riska attiecību. Veselības aprūpes speciālistus lūdz ziņot par visām iespējamām blakusparādībām, izmantojot valsts ziņošanas sistēmu. .
04.9 Pārdozēšana
Sunitiniba pārdozēšanai nav specifiska antidota, un pārdozēšanas ārstēšanā jāiekļauj vispārēji atbalstoši pasākumi. Ja nepieciešams, neabsorbētās aktīvās vielas elimināciju var veikt ar vemšanu vai kuņģa skalošanu. Par tiem ziņots. Pārdozēšanas gadījumi; dažos gadījumos šajos gadījumos radušās blakusparādības atbilda zināmajam sunitiniba drošības profilam.
05.0 FARMAKOLOĢISKĀS ĪPAŠĪBAS
05.1 Farmakodinamiskās īpašības
Farmakoterapeitiskā grupa: pretaudzēju līdzekļi, proteīnkināzes inhibitori.
ATĶ kods: LO1XE04.
Darbības mehānisms
Sunitinibs inhibē vairākus tirozīnkināzes (RTK) receptorus, kas ir iesaistīti audzēja augšanā, audzēja neoangioģenēzē un vēža metastātiskā progresēšanā. Sunitinibs ir identificēts kā trombocītu izcelsmes augšanas faktora receptoru (PDGFRα un PDGFRβ), asinsvadu endotēlija augšanas faktora receptoru (VEGFR1, VEGFR2 un VEGFR3) inhibitors, cilmes šūnu faktora receptors (KIT), FLT3 tirozīnkināzes receptors (inhibitors)Fms līdzīgs tirozīnkināze 3), CSF-1R receptori (koloniju stimulējošā faktora receptoru (CSF-1R)) un gliālās izcelsmes neitrofiskā faktora receptoru (RET). Galvenā metabolīta iedarbība bioķīmiskajos un šūnu testos ir līdzīga sunitiniba iedarbībai.
Klīniskā efektivitāte un drošība
Sunitiniba drošība un klīniskā efektivitāte ir pētīta, ārstējot pacientus ar GIST, kas ir rezistenti pret imatinibu (ti, tos pacientus, kuriem slimība ir progresējusi ārstēšanas laikā ar imatinibu vai pēc tās) vai nepanesību pret imatinibu (ti, tiem, kuriem ārstēšanas laikā bija nozīmīga toksicitāte). ar imatinibu, kas neļāva turpināt ārstēšanu), ārstējot pacientus ar MRCC un ārstējot pacientus ar neoperējamu pNET.
Efektivitātes pamatā ir laiks līdz audzēja progresēšanai un dzīvildzes palielināšanās pacientiem ar GIST, dzīvildze bez slimības progresēšanas un objektīvas atbildes reakcijas rādītājs iepriekš neārstētiem MRCC un MRCC pacientiem, kuri nav izturīgi pret terapiju ar MRCC, attiecīgi, citokīni, un pacientu dzīvildze bez progresēšanas ar pNET.
Kuņģa -zarnu trakta stromas audzēji (GIST)
Sākotnējais atklātais pieaugošais pētījums tika veikts pacientiem ar GIST pēc imatiniba neveiksmes (vidējā maksimālā dienas deva 800 mg) rezistences vai nepanesības dēļ. Tika iekļauti 97 pacienti ar dažādām devām un dozēšanas shēmām; 55 pacienti tika ārstēti ar SUTENT 50 mg saskaņā ar ieteicamo 4 nedēļu zāļu un divu nedēļu lietošanas pārtraukšanas grafiku (shēma 4/2).
Šajā pētījumā TTP vidējais laiks līdz progresēšanai bija 34,0 nedēļas (95% TI = 22,0-46,0 nedēļas).
Randomizēts, dubultmaskēts, placebo kontrolēts sunitiniba 3. fāzes pētījums tika veikts pacientiem ar GIST, kuri nepanesa slimību vai progresēja slimības laikā vai pēc ārstēšanas ar imatinibu (vidējā maksimālā dienas deva 800 mg). Šajā pētījumā 312 pacienti (2: 1) tika randomizēti, lai ārstētu ar 50 mg sunitiniba vai placebo iekšķīgi vienu reizi dienā saskaņā ar shēmu 4/2, līdz slimība progresē vai tiek pārtraukta no pētījuma cita iemesla dēļ (207 pacienti saņēma sunitinibu un 105 placebo).
Pētījuma primārais efektivitātes parametrs bija TTP, kas definēts kā laiks no nejaušināšanas brīža līdz pirmajai dokumentācijai par objektīvu audzēja progresēšanu. Iepriekš noteiktas starpposma analīzes laikā vidējais TTP ar sunitinibu bija 28,9 nedēļas (95% TI = 21,3 34,1 nedēļa), ja to novērtēja pētnieks, un 27,3 nedēļas (95% TI = 16,0–32,1 nedēļa), ja to novērtēja Neatkarīgā pārskata komiteja, un tas bija statistiski labāks par 5,1 nedēļu TTP, kas iegūts ar placebo (95% TI = 4,4–10,1) nedēļas, lpp
Neatkarīga pārskatīšanas komiteja. Kopējās dzīvildzes atšķirība statistiski bija par labu sunitinibam [bīstamības attiecība: 0,491 95% (TI 0,290-0,831)]; nāves risks bija 2 reizes lielāks pacientiem placebo grupā nekā sunitiniba grupā.
Pēc starpposma efektivitātes un drošības analīzes, pēc neatkarīgas DSMB ieteikuma, pētījums tika akls un pacientiem placebo grupā tika piedāvāts pāriet uz atklātu sunitiniba terapiju.
Pavisam pētījuma atklātajā ārstēšanas posmā ar sunitinibu tika ārstēti 255 pacienti, tostarp 99 pacienti sākotnēji tika ārstēti ar placebo.
Primārā un sekundārā parametra analīze pētījuma atklātajā posmā vēlreiz apstiprināja starpposma analīzes laikā iegūtos rezultātus, kā parādīts tabulā zemāk.
2. tabula. Efektivitātes parametru kopsavilkums (ITT populācija)
Vidējā kopējā dzīvildze (OS) ITT populācijā sunitiniba terapijas grupā un placebo grupā bija attiecīgi 72,7 nedēļas un 64,9 nedēļas (HR 0,876, 95% TI 0,679 - 1,129, p = 0,306). Šajā analīzē placebo grupā tika iekļauti tie pacienti, kuri tika randomizēti placebo un pēc tam pārgāja uz atklātu sunitiniba terapiju.
Metastātiska nieru šūnu karcinoma (MRCC) iepriekš neārstētiem pacientiem
Tika veikts nejaušināts, daudzcentru, starptautisks 3. fāzes pētījums, lai novērtētu sunitiniba efektivitāti un drošību salīdzinājumā ar IFN-α interferonu iepriekš neārstētiem MRCC pacientiem. Septiņi simti piecdesmit pacienti tika randomizēti 1: 1 pret ārstēšanas grupām; saņēma sunitiniba terapiju Atkārtoti 6 nedēļu cikli. Katrs cikls sastāv no 4 nedēļām ar 50 mg dienā perorāli, kam seko 2 nedēļas bez zāļu lietošanas (shēma 4/2), vai ar IFN-α, ko ievada iekšķīgi subkutāni 3 miljonu vienību devā. ) pirmajā nedēļā, 6 MV otrajā nedēļā un sākot no trešās nedēļas devā 9 MV, ārstējot 3 dienas pēc kārtas katru nedēļu.
Vidējais ārstēšanas ilgums bija 11,1 mēnesis (diapazons: 0,4 - 46,1) sunitiniba ārstēšanai un 4,1 mēnesis (diapazons 0,1 - 45,6) IFN -α ārstēšanai. Ar ārstēšanu saistītas nopietnas blakusparādības (TRSAE) tika ziņots 23,7% pacientu, kuri saņēma sunitinibu, un 6,9% pacientu, kuri saņēma IFN-α. Tomēr pārtraukšanas biežums blakusparādību dēļ bija 20% sunitiniba un 23% IFN-α. Ārstēšana tika pārtraukta 202 pacientiem (54%) sunitiniba grupā un 141 pacientam (39%) IFN-α grupā.
Devu samazināja 194 pacienti (52%), kuri tika ārstēti ar sunitinibu, un 98 pacienti (27%), kuri tika ārstēti ar IFN-α. Pacienti tika ārstēti līdz slimības progresēšanai vai pētījuma pārtraukšanai. Primārais efektivitātes kritērijs bija dzīvildze bez slimības progresēšanas (PFS).
Plānotā starpposma analīze parādīja statistiski nozīmīgu SUTENT priekšrocību salīdzinājumā ar IFN-α. Šajā pētījumā vidējais PFS sunitiniba grupai bija 47,3 nedēļas, salīdzinot ar 22,0 nedēļām IFN-α grupā; bīstamības attiecība bija 0,415 (95% TI: 0,320-0,539, p-vērtība
Ārstēšana ar sunitinibu bija saistīta ar ilgāku izdzīvošanu nekā ārstēšana ar IFN-α. Vidējā kopējā dzīvildze bija 114,6 nedēļas sunitiniba grupā (95% TI: 100,1–142,9 nedēļas) un 94,9 nedēļas IFN -α grupā (95% TI: 77,7–117,0 nedēļas) ar bīstamības attiecība no 0,821 (95% TI: 0,673-1,001; p = 0,0510, pamatojoties uz nestratificētu log-rank testu).
Izdzīvošana bez slimības progresēšanas (PFS) un kopējā dzīvildze (OS), kas novērota, lai ārstētu (ITT) populāciju un noteiktu ar radioloģisko laboratorijas novērtējumu, ir apkopota šādās tabulās:
Efektivitātes parametru kopsavilkums (ITT populācija)
Metastātiska citokīnu refraktāra nieru šūnu karcinoma (MRCC)
Otrās fāzes pētījums ar SUTENT tika veikts pacientiem, kuri nebija izturīgi pret iepriekšējo citokīnu terapiju un tika ārstēti ar interleikīnu-2 vai IFN-α. Sešdesmit trīs pacienti saņēma sākuma devu 50 mg sunitiniba perorāli vienu reizi dienā 4 nedēļas pēc kārtas, kam sekoja 2 nedēļu atpūtas periods, lai pabeigtu pilnu 6 nedēļu kursu (ārstēšanas shēma 4/2). Primārais efektivitātes parametrs bija objektīvās atbildes reakcijas rādītājs (Mērķis
Reakcijas ātrums (ORR)) saskaņā ar RECIST kritērijiem (Atbildes novērtēšanas kritēriji cietos audzējos).
Šajā pētījumā objektīvās atbildes reakcijas rādītājs bija 36,5% (95% TI 24,7% -49,6%), un vidējais laiks līdz progresēšanai (TTP) bija 37,7 nedēļas (95% TI 24,0-46,4 nedēļas).
Atklāts, vienas grupas, daudzcentru, apstiprinošs pētījums, lai novērtētu sunitiniba efektivitāti un drošību, tika veikts pacientiem ar MRCC, kas nav izturīgi pret iepriekšējo citokīnu terapiju. Simts seši pacienti saņēma vismaz vienu sunitiniba devu 50 mg. shēmas 4/2 ietvaros.
Šī pētījuma primārais efektivitātes parametrs bija ORR biežums. Sekundārie parametri ietvēra TTP, atbildes reakcijas ilgumu (DR) un kopējo dzīvildzi (OS).
Šajā pētījumā ORR bija 35,8% (95% TI 26,8% -47,5%). DR un vidējā OS vēl nebija sasniegta.
Aizkuņģa dziedzera neiroendokrīnie audzēji (pNET)
Atklātā daudzcentru, 2. fāzes atbalstošā pētījumā tika novērtēta sunitiniba monoterapijas efektivitāte un drošība, lietojot 50 mg dienas devas saskaņā ar 4/2 shēmu [4 ārstēšanas nedēļas, 2 nedēļas pārtraukuma] pacientiem ar neoperējamu pNET 66 pacientu grupā. pacientiem ar aizkuņģa dziedzera saliņu šūnu vēzi primārais atbildes reakcijas rezultāts bija 17%.
Tika veikts svarīgs 3. fāzes daudzcentru starptautisks randomizēts dubultmaskēts placebo kontrolēts pētījums par sunitiniba monoterapiju pacientiem ar neoperējamu pNET.
Pacienti, kuriem iepriekšējos 12 mēnešos bija nepieciešama dokumentēta slimības progresēšana, pamatojoties uz RECIST, tika randomizēti (1: 1), lai saņemtu 37,5 mg sunitiniba vienu reizi dienā bez plānotā atcelšanas perioda (n = 86) vai placebo (n = 85). .
Primārais mērķa kritērijs bija dzīvildzes bez slimības progresēšanas (PFS) novērtējums pacientiem, kuri lietoja sunitinibu, salīdzinot ar tiem, kuri saņēma placebo. Citi parametri bija OS, ORR procentuālā daļa, pacienta ziņotie rezultāti) un drošība.
No demogrāfisko īpašību viedokļa pacientu grupas, kas tika ārstētas ar sunitinibu un placebo, bija salīdzināmas. Turklāt 49% ar sunitinibu ārstēto pacientu un 52% pacientu, kuri saņēma placebo, bija nefunkcionējoši audzēji un 92% pacientu abās rokās bija metastāzes aknās. Pētījumā tika atļauts izmantot somatostatīna analogus. 66% pacientu, kuri tika ārstēti ar sunitinibu, un 72% pacientu, kuri tika ārstēti ar placebo, iepriekš tika ārstēti ar sistēmisku terapiju. Turklāt 24% pacientu sunitiniba grupā un 22% pacientu placebo grupā grupa saņēma somatostatīna analogus. Pētnieku novērtējumā tika novērota klīniski nozīmīga sunitiniba PFS priekšrocība salīdzinājumā ar placebo. Vidējais PFS bija 11,4 mēneši sunitiniba grupā, salīdzinot ar 5, 5 mēnešiem placebo grupā [riska attiecība: 0,418 (95% TI 0,263) , 0.662), p-vērtība = 0,0001]; līdzīgi rezultāti tika novēroti, novērtējot audzēja reakciju, pamatojoties uz RECIST pielietojumu izmeklētāju veiktajiem audzēja mērījumiem, lai noteiktu slimības progresēšanu, kā parādīts 3. tabulā. bīstamības attiecība par labu sunitinibam tika novērota visās apakšgrupās, kas tika novērtētas pēc sākotnējām īpašībām, ieskaitot analīzi pēc iepriekšējo sistēmisko terapiju skaita. Kopumā 29 pacienti sunitiniba grupā un 24 pacienti placebo grupā iepriekš nebija saņēmuši sistēmisku terapiju; šiem pacientiembīstamības attiecība PFS tas bija 0,365 (95% TI 0,156, 0,857), p = 0,0156.
Tāpat starp 57 pacientiem sunitiniba grupā (ieskaitot 28 pacientus ar 1 iepriekšēju sistēmisku terapiju un 29 ar 2 vai vairāk sistēmiskām terapijām) un 61 pacientu placebo grupā (ieskaitot 25 pacientus ar 1 iepriekšēju sistēmisku terapiju un 36 ar 2 vai vairāk sistēmiskām terapijām). ) l "bīstamības attiecība PFS tas bija 0,456 (95% TI 0,264, 0,787), p = 0,0036.
PFS jutīguma analīze tika veikta, ja PFS tika balstīta uz pētnieka audzēja mērījumiem un kad visi subjekti, kas tika cenzēti citu iemeslu dēļ, nevis pētījuma pārtraukšana, tika uzskatīti par PFS notikumiem. Šī analīze sniedza konservatīvu novērtējumu par sunitiniba terapijas ietekmi un apstiprināja primāro analīzi, parādot "bīstamības attiecība 0,507 (95% TI 0,350, 0,733), p = 0,000193. Galvenais pētījums par aizkuņģa dziedzera NET tika pārtraukts priekšlaicīgi, pamatojoties uz neatkarīgas Narkotiku novērtēšanas komitejas ieteikumu, un primārais parametrs bija balstīts uz pētnieka novērtējumu: abi apstākļi varēja ietekmēt ārstēšanas efekta novērtējumu.
Lai izslēgtu aizspriedumus pētnieku PFS novērtējumā, tika veikta neatkarīga, akla diagnostikas attēlveidošanas centrālā pārbaude; šis pārskats atbalstīja pētnieku novērtējumu, kā parādīts 3. tabulā.
3. tabula. Efektivitātes rezultāti no 3. fāzes pNET pētījuma
CI = ticamības intervāls, HR = riska attiecība, NA = nav piemērojams, NR = nav sasniegts
divpusējs log-rank nestrukturēts tests
b Fišera precīzais tests
Analīzes laikā kopējais dzīvildzes rādītājs nebija nobriedis. Sunitiniba grupā bija 9 nāves gadījumi, bet placebo grupā - 21 nāve. Statistiski nozīmīga atšķirība tika novērota sunitiniba objektīvās atbildes reakcijas salīdzinājumā ar placebo.
Slimības progresēšanas gadījumos pacienti tika informēti par saņemto ārstēšanu, un pacientiem, kuri lietoja placebo, tika piedāvāta iespēja iekļauties citā atklātā pagarinājuma pētījumā ar sunitinibu. Tā kā pētījums tika agri slēgts, pārējie pacienti tika informēti par ārstēšanu, ko viņi saņēma, un viņiem tika piedāvāta piekļuve atklātam pagarinātajam sunitiniba pētījumam. Kopumā 59 pacienti placebo grupā tika ārstēti ar sunitinibu. pagarinājumu.
Eiropas vēža izpētes un ārstēšanas organizācijas (EORTC QLQ-C30) aptaujas par dzīves kvalitāti rezultāti parādīja, ka vispārējā ar veselību saistītā dzīves kvalitāte un piecas funkcionālās jomas (fiziskā, lomu, kognitīvā, emocionālā un pacientiem, kuri saņēma sunitinibu, salīdzinot ar tiem, kuri saņēma placebo, saglabājās ar nelielām simptomātiskām blakusparādībām.
Pediatriskā populācija
Eiropas Zāļu aģentūra ir atlikusi pienākumu iesniegt SUTENT pētījumu rezultātus vienā vai vairākās pediatriskās populācijas apakšgrupās GIST ārstēšanai (informāciju par lietošanu bērniem skatīt 4.2. Apakšpunktā).
Eiropas Zāļu aģentūra ir atcēlusi pienākumu iesniegt pētījumu rezultātus ar SUTENT visās pediatriskās populācijas apakšgrupās nieru un nieru iegurņa karcinomu ārstēšanai (izņemot nefroblastomu, nefroblastomatozi, dzidru sarkomu šūnu, mezoblastisku nefromu, nieru medulāru). karcinoma un rabdoīds nieru audzējs) (informāciju par lietošanu bērniem skatīt 4.2. apakšpunktā).
Eiropas Zāļu aģentūra ir atbrīvojusi no pienākuma iesniegt pētījumu rezultātus ar SUTENT visās pediatriskās populācijas apakšgrupās gastroenteropankreātisko neiroendokrīno audzēju ārstēšanai (izņemot neiroblastomu, neiroganglioblastomu, feohromocitomu) (informāciju par lietošanu bērniem skatīt 4.2. ).
05.2 "Farmakokinētiskās īpašības
Sunitiniba farmakokinētiku novērtēja 135 veseliem brīvprātīgajiem un 266 pacientiem ar cietiem audzējiem. Farmakokinētika bija līdzīga visās pārbaudītajās populācijās ar cietu audzēju un veseliem brīvprātīgajiem.
25-100 mg devu shēmās laukums zem līknes (AUC) un Cmax palielinās proporcionāli devai. Atkārtoti lietojot katru dienu, sunitiniba uzkrājas 3-4 reizes, bet galvenais aktīvais metabolīts-7-10 reizes. Sunitiniba un tā galvenā aktīvā metabolīta līdzsvara koncentrācija tiek sasniegta 10-14 dienu laikā. Līdz 14. dienai sunitiniba un tā galvenā aktīvā metabolīta kopējā koncentrācija plazmā ir 62,9 - 101 ng / ml un atspoguļo paredzēto mērķa koncentrāciju, pamatojoties uz pirmsklīniskajiem datiem, lai kavētu receptoru fosforilēšanos. in vitro un noved pie audzēja stāzes / augšanas samazināšanās in vivo.
Galvenais aktīvais metabolīts veido 23–37% no kopējās zāļu iedarbības .. Atkārtotu reizi dienā vai atkārtotu kursu laikā, pārbaudot devu shēmas, nav novērotas būtiskas izmaiņas sunitiniba vai galvenā aktīvā metabolīta farmakokinētikā.
Uzsūkšanās
Pēc sunitiniba iekšķīgas lietošanas maksimālā koncentrācija (Cmax) parasti tiek novērota 6-12 stundu laikā (Tmax) pēc zāļu lietošanas.
Pārtika neietekmē sunitiniba bioloģisko pieejamību.
Izplatīšana
Sunitiniba un tā galvenā aktīvā metabolīta saistīšanās ar cilvēka plazmas olbaltumvielām testos iekšā in vitro tie bija attiecīgi 95% un 90% bez acīmredzamas atkarības no koncentrācijas.
Sunitiniba šķietamais izkliedes tilpums (Vd) bija liels - 2230 l - un tas norāda uz izplatīšanos audos.
Metabolisma mijiedarbība
Aprēķinātās Ki vērtības in vitro visām pārbaudītajām citohroma (CYP) izoformām (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4 / 5 un CYP4A9 / 11) norādīja, ka maz ticams, ka sunitinibs un tā galvenais aktīvais metabolīts klīniski nozīmīgā apjomā citu aktīvo vielu metabolismu, ko šie enzīmi var metabolizēt.
Biotransformācija
Sunitinibu galvenokārt metabolizē CYP3A4, citohroma P450 izoforma, kas ražo tā galveno aktīvo metabolītu desetilsunitinibu, ko tālāk metabolizē tas pats izoenzīms.
Jāizvairās no vienlaicīgas sunitiniba un spēcīgu CYP3A4 induktoru vai inhibitoru lietošanas, jo var mainīties sunitiniba līmenis plazmā (skatīt 4.4. Un 4.5. Apakšpunktu).
Eliminācija
Izvadīšana notiek galvenokārt ar izkārnījumiem (61%), un nemainītas aktīvās vielas un tās metabolītu eliminācija caur nierēm veido 16% no ievadītās devas. Sunitinibs un tā galvenais aktīvais metabolīts bija galvenie savienojumi, kas identificēti plazmā, urīnā un izkārnījumos un veidoja attiecīgi 91,5%, 86,4%un 73,8%no apvienotajos paraugos konstatētās radioaktivitātes. Nelieli metabolīti tika identificēti urīnā un izkārnījumos, bet parasti netika konstatēti plazmā. Kopējais klīrenss (CL / F) bija 34–62 l / h.
Pēc perorālas lietošanas veseliem brīvprātīgajiem sunitiniba un tā galvenā aktīvā metabolīta desetil eliminācijas pusperiods ir attiecīgi aptuveni 40–60 stundas un 80–110 stundas.
Īpašas populācijas
Aknu darbības traucējumiSunitinibu un tā galveno metabolītu galvenokārt metabolizē aknas. Sistēmiskā iedarbība pēc vienas sunitiniba devas pacientiem ar viegliem vai vidēji smagiem aknu darbības traucējumiem (Child-Pugh A un B stadija) bija līdzīga, salīdzinot ar indivīdiem ar normālu aknu darbību. SUTENT nav pētīts pacientiem ar smagiem aknu darbības traucējumiem (C pakāpe pēc Child-Pugh).
Vēža pētījumos netika iekļauti pacienti ar ALAT vai ASAT> 2,5 x NAR (augšējā normas robeža) vai ja šīs vērtības bija> 5,0 x NAR metastāžu dēļ.
Nieru darbības traucējumiPopulācijas farmakokinētikas analīzes liecināja, ka kreatinīna klīrenss attiecīgajā diapazonā (42-347 ml / min) neietekmēja šķietamo sunitiniba klīrensu (CL / F). Sistēmiskā iedarbība pēc vienas sunitiniba devas bija līdzīga pacientiem ar smagiem nieru darbības traucējumiem ( Lai gan sunitinibs un tā galvenais metabolīts netika izvadīti ar hemodialīzi pacientiem ar ESRD, kopējā sistēmiskā iedarbība bija par 47% mazāka sunitiniba un 31% tā galvenā metabolīta gadījumā, salīdzinot ar indivīdiem ar normālu nieru darbību.
Svars, veiktspējas statuss: Populācijas farmakokinētikas analīzes liecina, ka svara un Austrumu kooperatīvās onkoloģijas grupas (ECOG) darbības statusa pielāgošana nav nepieciešama.
Piederības dzimumsPieejamie dati liecina, ka sievietēm šķietamais sunitiniba klīrenss (CL / F) var būt par 30% mazāks nekā vīriešiem; tomēr šī atšķirība neprasa sākuma devas pielāgošanu.
05.3 Preklīniskie drošības dati
Atkārtotu devu toksicitātes pētījumi ar žurkām un pērtiķiem, kas ilga līdz 9 mēnešiem, parādīja, ka galvenais mērķorgānu efekts ir kuņģa -zarnu traktā (vemšana un caureja pērtiķiem); virsnieru dziedzeri (garozas sastrēgums un / vai asiņošana žurkām un pērtiķiem, ar nekrozi, kam seko žurku fibroze); asinis un limfātiskā sistēma (kaulu smadzeņu hipocelulārums un aizkrūts dziedzera, liesas un limfmezglu limfoīdais izsīkums); eksokrīnais aizkuņģa dziedzeris (acināru šūnu degranulācija ar vienšūnu nekrozi); siekalu dziedzeri (acināra hipertrofija); kaulu locītavas (augšanas plāksnes sabiezēšana); dzemde (atrofija) un olnīcas (folikulu attīstības samazināšanās). Visi rezultāti tika iegūti ar klīniski nozīmīgu sunitiniba koncentrāciju plazmā. Citi pētījumos novērotie efekti bija: QTc intervāla pagarināšanās, kreisā kambara izsviedes frakcijas (LVEF) un sēklinieku samazināšanās. cauruļveida atrofija, mezangiālo šūnu palielināšanās nierēs, kuņģa -zarnu trakta un mutes gļotādas asiņošana, hipofīzes priekšējo šūnu hipertrofija. endometrijs) un kaulu augšanas plāksne (epifīzes sabiezējums vai skrimšļa displāzija) ir saistīti ar sunitiniba farmakoloģisko iedarbību. Lielākā daļa no šiem notikumiem bija atgriezeniski pēc ārstēšanas pārtraukšanas uz 2-6 nedēļām.
Genotoksicitāte
Sunitiniba genotoksiskais potenciāls tika novērtēts in vitro un in vivo. Sunitinibs neuzrādīja mutagēnu iedarbību uz baktērijām, izmantojot žurku aknu nodrošināto metabolisko aktivāciju. Sunitinibs neizraisīja strukturālas hromosomu aberācijas cilvēka perifērajos limfocītos in vitro. Cilvēka perifērajos limfocītos ir novērota poliploīdija (hromosomu skaita novirzes) iekšā in vitro, gan metaboliskas aktivācijas klātbūtnē, gan bez tās. Sunitinibam žurku kaulu smadzenēs nebija klastogēna potenciāla in vivo. Galvenā aktīvā metabolīta potenciālais genotoksiskais potenciāls nav novērtēts.
Kancerogenitāte
Viena mēneša pētījumā, lai atrastu devu diapazonu (0, 10, 25, 75 vai 200 mg / kg / dienā), kurā rasH2 transgēnās peles baroja ar perorālu zondi, katru dienu un nepārtraukti ievadot ievadīto devu, plkst. tika novērotas lielākās devas (200 mg / kg / dienā) karcinoma un divpadsmitpirkstu zarnas Brunnera dziedzeru hiperplāzija.
Tika veikts 6 mēnešus ilgs pētījums, lai novērtētu kancerogenitāti rasH2 transgēnām pelēm, kuras tika barotas ar mutes caurulīti, ievadot ievadīto devu katru dienu (0, 8, 25, 75 [samazināts līdz 50] mg / kg dienā). Lietojot devas ≥25 mg / kg dienā 1 vai 6 mēnešus (≥ 7,3 reizes lielāks par AUC, kas novērots pacientiem, kuri saņēma ieteicamo dienas devu [RDD]), tika novērota kuņģa un divpadsmitpirkstu zarnas karcinoma, palielinājās sastopamības biežums. kuņģa gļotādu.
Divu gadu pētījumā, lai novērtētu kancerogenitāti žurkām (0, 0,33, 1 vai 3 mg / kg / dienā), lietojot sunitinibu 28 dienu ciklos, kam sekoja 7 dienu periodi bez devas, tika konstatēts biežuma pieaugums feohromocitomu un virsnieru dziedzera hiperplāziju žurku tēviņiem, kuri saņēma devu 3 mg / kg dienā ilgāk par 1 gadu (≥ 7,8 reizes pārsniedz AUC pacientiem, kuri saņēma RDD). Brunnera dziedzera karcinoma divpadsmitpirkstu zarnā radās sievietēm, lietojot ≥ 1 mg / kg dienā, vīriešiem - 3 mg / kg dienā, un gļotādas šūnu hiperplāzija bija redzama kuņģa dziedzeros vienādās devās, lietojot 3 mg / kg / kg. dienā vīriešiem; šie notikumi radās, lietojot 0,9, 7,8 un 7,8 reizes lielākas AUC pacientiem, kuri saņēma RDD. Šo neoplastisko atradumu, kas novēroti pelēm (rasH2 transgēna) un žurku kancerogenitātes pētījumos, nozīme cilvēkiem, kuri ārstēti ar sunitinibu, ir neskaidra.
Reproduktīvā un attīstības toksicitāte
Reproduktīvās toksicitātes pētījumos netika novērota ietekme uz vīriešu vai sieviešu auglību.Tomēr atkārtotu devu toksicitātes pētījumi ar žurkām un pērtiķiem parādīja ietekmi uz mātīšu auglību folikulu atrēzijas, dzeltenā ķermeņa deģenerācijas, dzemdes endometrija izmaiņu un dzemdes un olnīcu svara samazināšanās veidā līdz klīniski nozīmīgam līmenim. sistēmiska iedarbība. Ietekme uz žurku tēviņu auglību tika novērota sēklinieku cauruļveida atrofijas, spermatozoīdu samazināšanās epididimā un koloidālā izsīkuma dēļ prostatā un sēklas pūslīšos, ja plazmas iedarbības līmenis 25 reizes pārsniedza cilvēka sistēmisko iedarbību.
Žurkām embrija-augļa mirstība izraisīja dzīvu augļu skaita ievērojamu samazināšanos, palielinātu rezorbciju skaitu, palielinātu zaudējumu skaitu pēc implantācijas un kopējos pēcnācēju zaudējumus 8 no 28 grūtniecēm, ja iedarbības līmenis plazmā bija 5,5 reizes lielāks nekā sistēmiskā iedarbība uz cilvēkiem . Trušiem grūtnieces dzemdes svara un dzīvo augļu skaita samazināšanos izraisīja augļa rezorbciju pieaugums, zaudējumi pēc implantācijas un kopējie pēcnācēju zaudējumi 4 no 6 grūtniecēm, ja plazmas ekspozīcijas līmenis bija 3 reizes lielāks. cilvēkos. Ārstēšana ar sunitinibu žurkām organoģenēzes laikā deva ietekmi uz attīstību, lietojot devas ≥ 5 mg / kg / dienā, kas palielināja augļa skeleta anomāliju sastopamību, ko galvenokārt raksturo aizkavēta krūšu kurvja / jostas skriemeļu ossifikācija un kas rodas pie plazmas iedarbības līmeņa 5.5 reizes cilvēka sistēmiskā iedarbība. Trušiem ietekme uz attīstību ietvēra palielinātu lūpu plaisas sastopamību plazmā, kas bija aptuveni tāda pati kā klīnikā, un lūpu un aukslēju šķeltnes, ja plazmas iedarbības līmenis 2,7 reizes pārsniedza sistēmisko iedarbību cilvēkam.
Sunitiniba (0,3; 1,0; 3,0 mg / kg / dienā) ietekme uz pirms un pēcdzemdību attīstību tika novērtēta pētījumā ar žurku mātītēm. Mātes ķermeņa masas palielināšanās grūtniecības un zīdīšanas laikā tika samazināta, lietojot devas> 1 mg / kg / dienā, bet līdz 3 mg / kg / dienā devām netika novērota toksiska ietekme uz reproduktīvo funkciju (aprēķinātā iedarbība> 2, 3 reizes pārsniedza AUC, kas konstatēta pacientiem samazināts pēcnācēju ķermeņa svars, lietojot devas 3 mg / kg dienā pirms un pēc atšķiršanas. Netika novērota toksiska ietekme uz attīstību, lietojot devas 1 mg / kg dienā (iedarbība aptuveni ≥ 0,9 reizes lielāka par AUC novērots pacientiem, kuri saņem RDD).
06.0 FARMACEITISKĀ INFORMĀCIJA
06.1 Palīgvielas
Kapsulas saturs
Mannīts (E421)
Nātrija kroskarmeloze
Povidons (K-25)
Magnija stearāts
Kapsulas apvalks
Želeja
Sarkanais dzelzs oksīds (E172)
Titāna dioksīds (E171)
Tinte
Šellaks
Propilēnglikols
Nātrija hidroksīds
Povidons
Titāna dioksīds (E171)
06.2 Nesaderība
Nav būtisks.
06.3 Derīguma termiņš
3 gadi.
06.4 Īpaši uzglabāšanas nosacījumi
Šīm zālēm nav nepieciešami īpaši uzglabāšanas apstākļi.
06.5 Tiešā iepakojuma veids un iepakojuma saturs
Augsta blīvuma polietilēna (ABPE) pudeles ar polipropilēna aizbāzni, kas satur 30 cietās kapsulas.
Caurspīdīgi poli (hlorotrifluoretilēna) / PVC blisteri ar perforētām vienības devām un alumīnija foliju ar termiski noslēgtu laku, kas satur 28 x 1 cietās kapsulas.
Visi iepakojuma lielumi tirgū var nebūt pieejami.
06.6 Norādījumi lietošanai un lietošanai
Nav īpašu norādījumu.
07.0 REĢISTRĀCIJAS APLIECĪBAS ĪPAŠNIEKS
SIA Pfizer
Ramsgeitas ceļš
Sviestmaize, Kent CT13 9NJ
Lielbritānija
08.0 REĢISTRĀCIJAS APLIECĪBAS NUMURS
EU/1/06/347/001
037192022
EU/1/06/347/004
09.0 PIRMĀJAS APLIECĪBAS VAI ATĻAUJAS DATUMS
Reģistrācijas datums: 2006. gada 19. jūlijs
Pēdējās pārreģistrācijas datums: 2012. gada 9. janvāris
10.0 TEKSTA PĀRSKATĪŠANAS DATUMS
11.0 RADIOZĀLĒM, PILNĪGI DATI PAR IEKŠĒJO RADIĀCIJAS DOSIMETRIJU
12.0 RADIOZĀLĒM, PAPILDU SĪKĀKI NORĀDĪJUMI PAR ĪSTENAJU SAGATAVOŠANU UN KVALITĀTES KONTROLI